












ПРИМЕНЕНИЕ БОТОКСА ГРОЗИТ
РАЗВИТИЕМ ТЯЖЕЛЫХ ОСЛОЖНЕНИЙ
 Ботокс далеко не безопасен, предупреждают эксперты Университета Висконсина (США). Они доказали, что после введения в организм ботулотоксин распространяется по всему организму и может нанести серьезный вред внутренним органам.
Ботокс далеко не безопасен, предупреждают эксперты Университета Висконсина (США). Они доказали, что после введения в организм ботулотоксин распространяется по всему организму и может нанести серьезный вред внутренним органам.
25.10.2020 г.
Степень распространения ботулотоксина напрямую зависит от объема введенной дозы. До сих пор ученые полностью не изучили данный процесс. Действительно, есть основания полагать, что молекулы Ботокса способны перемещаться между нейронами.
В доказательство приводится эксперимент с грызунами. Итак, частично ботулотоксин действовал местно, но частично распространился по телу, что повышает риск внезапного паралича. Молекулы препарата используют для перемещения аксоны – длинные отростки нервных клеток, по которым идут нервные импульсы. Несмотря на заявления ученых, официально считается, что Ботокс крайне редко вызывает побочные эффекты.
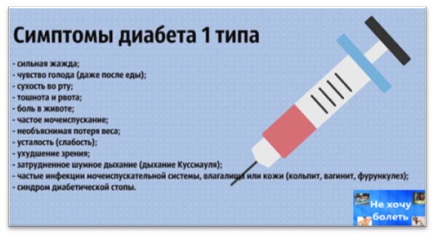
ПРИЕМ АНТИБИОТИКОВ В ДЕТСТВЕ
ПРИВОДИТ К ДИАБЕТУ 1 ТИПА
Антибиотики часто назначаются в качестве перестраховки, когда они не нужны, что грозит развитием диабета 1-го типа. Диабет 1-го типа относится к аутоиммунным заболеваниям. Его обычно диагностируют в детстве. По мнению врачей и ученых, здесь играет роль генетика, и изменение микробиома.
23.10.2020 г.
Но есть и внешний фактор. Об этом говорит тот факт, что количество диабетиков (и много маленьких детей) значительно выросло после Второй мировой, когда антибиотики вошли в повседневную жизнь.
Ученые взяли группу мышей с диабетом и дали тем одну из двух дозировок антибиотиков (или очень низкую дозу, что дают животным на фермах, или высокую, прописываемую детям при инфекциях горла). При этом у самцов, как выяснили специалисты, воздействие большей дозы антибиотиков в первые дни жизни усиливало развитие диабета. А вот малая доза антибиотиков не приводила к этому заболеванию.
А микробиом (бактериальный состав кишечника) важен для метаболизма и иммунитета. Любые аномальные изменения микробиома грозят развитием недугов. Более того, были зафиксированы изменения в экспрессии генов в кишечнике мышей. Отмечались также негативные сдвиги в метаболизме (касались всех генов, задействованных в метаболизме холестерина).
meddaily.ru
ТЕСТ СТОИМОСТЬЮ В $1 ОБНАРУЖИТ
ПОДДЕЛЬНЫЕ ЛЕКАРСТВА
Люди, приобретающие лекарства в интернет-аптеках, рискуют купить поддельный препарат или же лекарственное средство, которое неправильно хранилось и потому может действовать не так, как заявлял производитель. Внешне же упаковка может выглядеть неповрежденной, а фальсификат может продаваться в такой же пачке, как и настоящее лккаство. Особенно остро эта проблема стоит в развивающихся странах.

22.10.2020 г.
Для того чтобы уберечь потребителя от приобретения поддельного лекарства, фармпроизводители и власти придумывают разные способы.
Американские ученые из Университета Нотр-Дам (University of Notre Dame) создали недорогой и простой в использовании тест, который позволит определить, соответствует ли состав лекарственного средства заявленному. Стоимость бумажной тест-полоски – 1 доллар.
Каждая тест-система на бумажной основе содержит 12 индикаторов, распознающих те или иные компоненты или функциональные группы соединений, входящих в состав препаратов. Порядок проведения анализа следующий: тестируемую таблетку нужно измельчить, полученные порошок нанести на 12 индикаторных «дорожек», затем поместить полоску в воду на 3 минуты. После нужно сравнить окраску «дорожек» с контрольным образцом. Для этого можно воспользоваться специально разработанным приложением, но можно обойтись и без него, сравнивая цветвизуально.
Существующая сейчас тест-система предназначена для оценки состава распространенных антибиотиков. В будущем исследователи планируют создать тест-полоски и для обнаружения других поддельных лекарств.
Эффективность тест-системы была проверена на образцах, полученных из Кении и Уганды. Лекарства в странах Африки часто хранятся при неправильном температурном режиме, а их транспортировка проходит с нарушениями. Ученые проверили состав лекарств с помощью своей разработки и с использованием масс-спектрометрии. Им удалось выявить фальсификат, а также лекарства, разрушившиеся под действием высоких температур.
doctorpiter.ru
FDA ОБНОВЛЯЕТ ИНФОРМАЦИЮ
О ПРОФИЛЕ БЕЗОПАСНОСТИ ФТОРХИНОЛОНОВ
Управление по контролю за пищевыми продуктами и лекарственными средствами США (FoodandDrugAdministration – FDA) одобрило изменения маркировки относительно профиля безопасности одного из класса антибиотиков (фторхинолонов) с целью предупреждения о возможной связи между их применением и потерей трудоспособности, а также потенциально долговременных побочных эффектов и необходимости ограничения их применения у пациентов с менее серьезными бактериальными инфекциями.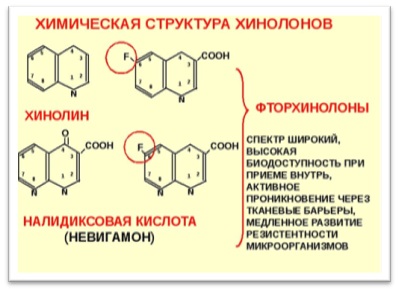
22.09.2020 г.
Фторхинолоны – антибиотики, которые убивают или останавливают рост бактерий. Хотя эти препараты эффективны при лечении тяжелых бактериальных инфекций, FDA установлено, что применение как оральных, так и инъекционных форм фторхинолонов связано с потерей трудоспособности ввиду наличия побочных эффектов, которые негативно влияют на сухожилия, мышцы, суставы и центральную нервную систему. Эти побочные эффекты могут возникать спустя несколько часов или несколько недель после применения фторхинолонов и быть долговременными.
Поскольку риск возникновения этих серьезных побочных эффектов, как правило, перевешивает пользу для пациентов с острым бактериальным синуситом, обострением хронического бронхита и неосложненными инфекциями мочевыводящих путей, FDA установило, что фторхинолоны должны применяться у пациентов с данными заболеваниями при отсутствии альтернативных вариантов лечения. Для некоторых серьезных бактериальных инфекций, включая сибирскую язву, чуму и бактериальную пневмонию, польза от применения фторхинолонов перевешивает риски, поэтому данный класс антибиотиков остается вариантом их терапевтического лечения.
Одобренные FDA фторхинолоны: левофлоксацин, ципрофлоксацин (в том числе в форме таблеток пролонгированного действия), моксифлоксацин, офлоксацин и гемифлоксацин. Изменения маркировки данных антибиотиков включают обновление информации на упаковке, а также в инструкции в разделе «Предупреждения и меры предосторожности об опасности возникновения нетрудоспособности и потенциально необратимых побочных реакций, которые могут происходить одновременно». На этикетке также будет содержаться информация об ограничении применения фторхинолонов у пациентов с острым бактериальным синуситом, обострением хронического бронхита и неосложненными инфекциями мочевыводящих путей, не имеющих других доступных вариантов лечения.
FDA впервые добавило предупреждение относительно повышенного риска развития тендинита и разрыва сухожилия при применении фторхинолонов на упаковке препаратов в июле 2008 г. В феврале 2011 г. на упаковки данного класса антибиотиков также была добавлена информация о возможном риске обострения симптомов у лиц с миастенией. В августе 2013 г. FDA потребовало обновления маркировки фторхинолонов для добавления информации о возможном развитии необратимой периферической нейропатии при их применении.
В ноябре 2015 г., Консультативный комитет FDA обсудил риски и пользу при применении фторхинолонов для лечения острого бактериального синусита, обострения хронического бронхита и неосложненных инфекций мочевыводящих путей на основе новой информации о профиле безопасности этих антибиотиков, которая была сосредоточена на 2 или более побочных эффектах, происходящих одновременно и вызывающих потенциально необратимые нарушения. Консультативный комитет пришел к выводу, что серьезные риски, связанные с применением фторхинолонов при вышеуказанных видах неосложненных инфекций, как правило, перевешивает пользу для пациентов с другими вариантами лечения.
Впоследствии 12 мая 2016 г. FDA пришло к выводу о необходимости указания информации о том, что фторхинолоны следует применять при данных заболеваниях только в случае отсутствия других доступных вариантов лечения из-за потенциально опасных долгосрочных побочных эффектов при их применении, на маркировке антибиотиков данного класса.
«Применение фторхинолонов имеет свои риски и выгоды, которые следует рассматривать очень внимательно», – отметил Эдвард КОКС (Edward COX), врач, директор Управления антимикробных продуктов в центре FDA по оценке и исследованию препаратов (AntimicrobialProductsintheFDA’sCenterforDrugEvaluationandResearch). – Важно, чтобы медработники и пациенты знали, каковы риски и преимущества фторхинолонов, и принимали обоснованное решение об их применении».
fda.gov
Фото: myslide.ru
NICE НЕ РЕКОМЕНДУЕТ ПРЕПАРАТ «НЕКСАВАР» (СОРАФЕНИБ)
ДЛЯ ЛЕЧЕНИЯ РАКА ПЕЧЕНИ
 Национальный институт здоровья Великобритании (NICE) не одобрил использование лекарственного препарата «Нексавар» (Nexavar, сорафениб/sorafenib) немецкой фармацевтической компании немецкой Bayer для лечения рака печени, так он не соответствует стандартам соотношения «цена-качество».
Национальный институт здоровья Великобритании (NICE) не одобрил использование лекарственного препарата «Нексавар» (Nexavar, сорафениб/sorafenib) немецкой фармацевтической компании немецкой Bayer для лечения рака печени, так он не соответствует стандартам соотношения «цена-качество».
20.09.2020 г.
NICE и ранее не поддержал использование лекарственного средства в государственном здравоохранении для лечения рака печени по этой же причине. Компания уменьшила стоимость препарата. Однако специалисты NICE заявили, что лекарственное средство будет рекомендовано только при очевидной пользе и экономической эффективности для здравоохранения страны, а на данный момент нет определенности относительно выживаемости в целом, и неясно, как долго пациенты будут принимать лекарство.
Компании Bayer было предложено предоставить NICE дополнительную информацию о Нексавар (сорафениб).
FDA ПРЕДУПРЕДИЛА О СВЯЗИ ИНГИБИТОРОВ SGLT2 И РАЗВИТИИ ТЯЖЕЛЫХ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ

30 августа 2018
Американские контрольные органы предупредили о повышенном риске развития тяжелых форм мочеполовых инфекций при применении противодиабетических препаратов класса ингибиторов SGLT2. По данным FDA, было зарегистрировано 12 подобных случаев, 11 из них привели к госпитализации пациентов, а один – к летальному исходу. Об этом сообщает Reuters.
Речь идет о гангрене Фурнье – редкой жизнеугрожающей бактериальной инфекции, поражающей мягкие ткани вокруг гениталий, говорится в заявлении FDA. За пять лет были получены сообщения о 12 случаях гангрены Фурнье (7 – у мужчин и 5 – у женщин). Инфекция развилась в течение нескольких месяцев после начала терапии ингибиторами SGLT2.
Ингибиторы натрийглюкозного котранспортера 2 типа (SGLT2) являются высокоэффективными средствами терапии диабета 2 типа. Подавляя активность SGLT2, данные препараты уменьшают реабсорбцию прошедшей фильтрацию глюкозы, тем самым повышая ее выведение почками. Первое лекарственное средство данного класса было зарегистрировано в США в 2013 году. В настоящее время на рынке представлены ингибиторы SGLT2 производства Johnson & Johnson, Eli Lilly, Bristol-Myers Squibb, Astra Zeneca, MSD и Pfizer.
Дополнение от компании AstraZeneca
29 августа FDA выпустила заявление по результатам проведенного анализа базы данных отчетов по нежелательным и серьезным нежелательным явлениям, суть которого заключается в том, что при использовании у пациентов с СД2 препаратов класса SGLT2i в редких случаях наблюдалось развитие редкого заболевания инфекционного генеза – гангрены Фурнье.
После получения уведомления от FDA об изучении данного вопроса AstraZeneca проанализировала данные из всех источников с начала этого года относительно редких случаев возникновения гангрены Фурнье и пришла к заключению о невозможности установления причинно-следственной связи между использованием дапаглифлозина и возникновением гангрены Фурнье.
«Безопасность пациентов является ключевым приоритетом для компании AstraZeneca, и мы всегда придерживаемся правила передавать всю необходимую информацию о безопасности наших препаратов медицинским работникам, соответствующим органам здравоохранения и пациентам», – заявили в компании.
remedium.ru
ПОВЫШЕНИЕ ЧАСТОТЫ РЕЦИДИВА ОНКОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ У ПАЦИЕНТОВ, ПОЛУЧАЮЩИХ АЗИТРОМИЦИН ПОСЛЕ ТРАНСПЛАНТАЦИИ СТВОЛОВЫХ КЛЕТОК
 15 августа 2018
15 августа 2018
FDA сообщает об отрицательных последствиях длительной терапии азитромицином у пациентов с раком крови и лимфатических узлов, которые перенесли донорскую трансплантацию стволовых клеток.
Согласно результатам исследования, опубликованного Bergeron A. с соавторами в JAMA в 2017 году, длительная терапия у таких пациентов повышает риск рецидива опухоли. Эксперты FDA провели подробный анализ имеющихся данных.
У пациентов, перенесших трансплантацию донорских стволовых клеток, повышен риск развития синдрома облитерирующего бронхиолита – заболевания, которое сопровождается воспалением и образованием рубцов в дыхательных путях и легких и проявляется сухим кашлем и выраженной одышкой. Хотя азитромицин и не одобрен для профилактики синдрома облитерирующего бронхиолита, антибиотик активно назначается таким пациентов.
Исследование ALLOZITHRO
Исследование ALLOZITHRO, послужившее поводом для обзора безопасности FDA, изучало эффективность и безопасность терапии азитромицином (3 раза в неделю по 250 мг), назначаемой на протяжении 2 лет. Важно отметить, что исследование было завершено после 13 месяцев по причине повышенного риска рецидива рака и смерти в группе азитромицина.
Рецидив рака наблюдался у 32,9% на фоне азитроимицина и 20,8% на фоне плацебо. Частота 2-летней выживаемости составила 56,6% и 70,1% соответственно. Обращало на себя внимание, что в первые месяцы исследования смертность была сравнимой в двух группах, но постепенно между группами были выявлены достоверные различия, свидетельствующие о низкой безопасности азитромицина.
Источники:
FDA warns about increased risk of cancer relapse
with long-term use of azithromycin antibiotic after donor stem cell transplant, 3 August 2018.
Bergeron A, Chevret S, Granata A, et al. JAMA 2017;318(6):557-566.
FDA ОБНОВИЛО ДАННЫЕ ПО БЕЗОПАСНОСТИ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ
 30 августа 2018
30 августа 2018
Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) обновило информацию по безопасности лекарственных препаратов.
Элуксадолин: реакции гиперчувствительности и анафилактические реакции.
Аотбутерол, ипратропий+альбутерол: серьезные кожные реакции.
Токлизумаб, ринолацепт, канакинумаб, сарилумаб, анакинра: ингибиторы ИЛ-1 и ИЛ-6 повышают риск развития легочной гипертензии, интерстициальной болезни легких, легочного альвеолярного протеиноза.
Риоцигуат: обморок.
Золпидем, эсзопиклон, залеплон: сомнамбулизм, анормальные, ассоциированные со сном реакции.
Босутиниб, иматиниб, пронатиниб, дазатиниб, нилотиниб: тромботическая микроангиопатия.
Левокарнитин: реакции гиперчувствительности и анафилактические реакции.
Шприц-ручка с инсулином (инсулин глулизин, инсулин гларгин, инсулин аспарт, инсулин лиспро, инсулин детемир): ошибка в использовании препарата, пациенты забывают открыть внутренней крышки игл.
Синкалид: анафилактический шок и другие реакции гиперчувствительности.
Позаконазол, итраконазол: повышение минералкортикоидной активности.
Пропилтиоурацил: фатальные случаи АНЦА-ассоциированного васкулита.
Родапилант (таблетки и раствор для инъекций): реакции гиперчувствительности и реакции, ассоциированы с инфузией препарата.
Источник:
Marcia Frellick. New FDA Watch List Has Insulin Pens,
Muscle Relaxants, and More. Medscape. July 31, 2018
FDA ОГРАНИЧИЛО ПРИМЕНЕНИЕ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ «КЕЙТРУДА» И «ТЕЦЕНТРИК»
4 сентября 2018
FDA ограничило применение препаратов «Кейтруда» фармацевтической компании Merck (за пределами США и Канады – MSD) и «Тецентрик» компании Roche у пациентов, страдающих местно-распространенным или метастатическим раком мочевого пузыря, которым невозможно проведение химиотерапии препаратами платины.
Теперь использование препарата «Кейтруда» разрешено только у пациентов из этой группы, у которых экспрессия опухолями биомаркера PD-L1 соответствует совокупной положительной оценке 10 баллов или выше. Препарат «Тецентрик» может назначаться только тем пациентам, у которых иммунные клетки PD-L1 составляет минимум 5% от всей площади опухоли.
Данное решение регулятора основано на результатах клинических исследований, согласно которым у пациентов с низким уровнем PD-L1, принимавших Кейтруда или Тецентрик, уменьшались показатели выживаемости по сравнению с теми, кто получал химиотерапию препаратами платины.
ЕВРОПЕЙСКИЕ СТРАНЫ ОТЗЫВАЮТ ОПАСНЫЙ КАРДИОЛОГИЧЕСКИЙ ДЖЕНЕРИК, ПРОИЗВЕДЕННЫЙ В КИТАЕ
 5 июля 2018 года появилась информация, что европейские страны отзывают ряд препаратов для лечения артериальной гипертензии, которые содержат опасный компонент, произведенный в Китае. Опасное вещество обвиняют в канцерогенном эффекте.
5 июля 2018 года появилась информация, что европейские страны отзывают ряд препаратов для лечения артериальной гипертензии, которые содержат опасный компонент, произведенный в Китае. Опасное вещество обвиняют в канцерогенном эффекте.
История вопроса. Европейское медицинское агентство инициировало расследование в связи с обнаружением примеси N-нитрозодиметиламин (НДМА) в активном веществе валсартана. Все подозрительней препараты были произведены китайской компанией Zhejiang Huahai Pharmaceuticals. Предполагают, что опасная примесь появилась в валсартане после изменения процесса получения лекарства. Важно отметить, что НДМА представляет собой потенциальный человеческий канцероген.
В то время как ЕМА проводит расследование безопасности (изучает уровень НДМА в валсартане и возможные последствия для пациентов, которые принимали препарат), национальные фармакологические комитеты в Европе отзывают партии валсартана, произведенные Zhejiang Huahai.
Как известно, валсартан часто используется для лечения пациентов с артериальной гипертензией для снижения сердечно-сосудистого риска (в первую очередь, инсульта и инфаркта), а также у пациентов с сердечной недостаточностью и тех, кто недавно перенес сердечный приступ.
Источник:
EMA reviewing medicines containing valsartan from Zhejiang Huahai
following detection of an impurity, 05 July 2018
ПРОИЗВОДИТЕЛЬ ОСТАНОВИЛ РЕАЛИЗАЦИЮ ЛЕКАРСТВА ДЛЯ ГИПЕРТОНИКОВ ИЗ-ЗА НЕИЗВЕСТНЫХ ПРИМЕСЕЙ
 Компания «Гедеон Рихтер» решила приостановить продажу сразу одиннадцати серий лекарства для гипертоников, содержащего валсартан. Такое решение производитель принял из-за того, что другая компания нашла в этом действующем веществе неизвестные примеси.
Компания «Гедеон Рихтер» решила приостановить продажу сразу одиннадцати серий лекарства для гипертоников, содержащего валсартан. Такое решение производитель принял из-за того, что другая компания нашла в этом действующем веществе неизвестные примеси.
Представитель ОАО «Гедеон Рихтер» (Венгрия) сообщил Росздравнадзору о решении приостановить реализацию препарата «Нортиван, таблетки, покрытые пленочной оболочкой, 80 мг» серий H5B033D, H5B034A, H69139A, H79073E и в дозировке 160 мг серий H58026A, H5A002B, H5A004A, H5C078A, H75052A, H79107A, H81091A.
Такое решение фармкомпания приняла из-за того, что китайский производитель Чжэцзян Хуахай Фармасьютикал Ко.Лтд. провел экспертизу и обнаружил в фармацевтической субстанции валсартан неизвестную примесь. Это же действующее вещество содержится и в «Нортиване».
Лекарство «Нортиван» обладает антигипертензивными свойствами. Снижает повышенное артериальное давление и назначается для лечения артериальной гипертензии и хронической сердечной недостаточноcти.
Ранее Доктор Питер писал том, как фармкомпании обратили внимание на рекомендации Комитета по оценке рисков Европейского агентства лекарственных средств (PRAC/EMA): в начале года эксперты выяснили, что из-за обезболивающих, содержащих флупиртин, у пациентов могут появиться проблемы с печенью. Зарубежные фармпроизводители отозвали анальгетики из-за небезопасного действующего вещества, однако российские производители международные рекомендации проигнорировали.
doctorpiter.ru
ПОВЫШЕНИЕ СЕРДЕЧНО-СОСУДИСТОГО РИСКА НА ФОНЕ ТЕРАПИИ НПВП
Актуальность. Нестероидные противовоспалительные препараты (НПВП) составляют основу терапии пациентов с поражением суставов и связочного аппарата. При этом их прием ассоциирован с многочисленными рисками, в том числе повышением риска сердечно-сосудистых заболеваний более, чем в 2 раза. Новые данные о безопасности НПВП были представлены на конгрессе European League Against Rheumatism (EULAR) в Амстердаме.
Дизайн и результаты исследования. Канадские исследователи включили в анализ 7 743 пациента с остеоартритом и сопоставили их по возрасту и полу с 23 229 индивидуумами, не имеющими данного заболевания.
Мультивариантный анализ показал, что риск развития впервые диагностированных сердечно-сосудистых заболеваний был на 23% выше у пациентов с остеоартритом, с учетом возраста, пола, социо-экономического статуса, ИМТ, гипертонии, сахарного диабета, гиперлипидемии, ХОБЛ.
Обращало на себя внимание, что риск сердечной недостаточности в группе больных остеоартритом был на 42% выше, чем в контрольной группе, риск ИБС – на 17%, риск инсульта – на 14% выше.
Анализ показал, что НПВП вносили 67,5% риска в развитие сердечно-сосудистых заболеваний у пациентов с остеоартритом. В частности, риск сердечной недостаточности увеличивался на 44,8% при использовании НПВП, риск ИБС – на 94,5%, риск инсульта – на 93,3%.
Комментируя результаты исследования, авторы отмечают, что применение НПВП у больных с остеоартритом и сердечно-сосудистые риски представляют собой порочный круг: не существует альтернативы НПВП, но вместе с тем нежелательные явления НПВП со стороны сердца и почек могут наносить значительный вред пациентам.
Источник:
European League Against Rheumatism (EULAR) Congress 2018: Abstract OP0190.
Presented June 14, 2018.
РИСК РАЗВИТИЯ ЛЕКАРСТВЕННО УСТОЙЧИВЫХ БАКТЕРИЙ У ПАЦИЕНТОВ С АЛЛЕРГИЕЙ НА ПЕНИЦИЛЛИН
 Известно, что излишнее использование антибиотиков широкого спектра приводит к развитию лекарственно резистентных бактерий, включая метициллин резистентного Staphylococcus aureus (MRSA) и внутрибольничной инфекции Clostridium difficilе. Исследователи оценили связь между аллергией на пенициллин и развитием MRSA и инфекции Clostridium difficilе.
Известно, что излишнее использование антибиотиков широкого спектра приводит к развитию лекарственно резистентных бактерий, включая метициллин резистентного Staphylococcus aureus (MRSA) и внутрибольничной инфекции Clostridium difficilе. Исследователи оценили связь между аллергией на пенициллин и развитием MRSA и инфекции Clostridium difficilе.
Актуальность. Треть пациентов отмечают аллергию на лекарства (нежелательные или аллергические реакции). Препаратами с наибольшим числом аллергий считаются пенициллины (от 5 до 16%). У пациентов с аллергией на пенициллин часто назначаются антибиотики более широкого спектра и обладающие более высокой токсичностью.
Дизайн исследования. За основу было взято популяционное когортное исследование, проводимое в кабинетах Великобритании с 1995 по 2015 годы.
В исследование вошли 301 399 взрослых без указания в анамнезе на MRSA или C difficile, из них у 64 141 имела место аллергия на пенициллин и 237 258 индивидуумов вошли в группу контроля (сопоставлены по возрасту, полу и дате включения в исследование).
Первичной конечной точкой исследования являлась частота развития MRSA и C difficile. Вторичной – частота использования β-лактамных антибиотиков и альтернативных антибиотиков.
Результаты. Средний период наблюдения за участниками исследования составил 6 лет.
Среди 64 141 взрослого пациента с аллергией на пенициллин и 237 258 индивидуумов из группы сравнения у 1 365 был диагностирован MRSA (442 пациента из группы аллергии на пенициллин и 923 – из контрольной группы) и у 1 688 развилась инфекция C difficile (442 и 1 246 индивидуума соответственно).
Среди пациентов с аллергией на пенициллин скорректированный коэффициент рисков для MRSA был равен 1,69 (95% CI, l 1,51-1,90) и для C difficile 1,26 (95% CI, 1,12-1,40).
Относительный риск использования антибиотиков среди пациентов с аллергией на пенициллин составил 4.15 (95% CI, 4,12-4,17) для макролидов, 3,89 (95% CI, 3,66-4,12) для клиндамицина и 2,10 (95% CI, 2,08-2,13) для фторхинолонов. Использование альтернативных β-лактамных антибиотиков повышало риска развития MRSA на 55%, а риск C difficile на 35%.
Заключение. Аллергия на пенициллин ассоциирована с повышением риска развития MRSA и C difficile, что опосредованно увеличением применения альтернативных β-лактамам антибиотиков.
Источник:
Kimberly G Blumenthal, Na Lu, Yuqing Zhang, et al.
BMJ 2018;361:k2400,
internist.ru
ОСОБЕННОСТИ ЭКСПЕРТИЗЫ МЕДИЦИНСКИХ ИЗДЕЛИЙ ДЛЯ IN VITRO ДИАГНОСТИКИ В КАЗАХСТАНЕ
Медицинские изделия для диагностики in vitro ни при каких условиях не должны контактировать с организмом пациента, то есть они ни при каких условиях не могут оказать воздействия на организм, и, соответственно, государственная экспертиза таких изделий должна проводиться на самом высокой уровне. Об этом рассказывает Павел Николаевич ДЕРЯБИН, доктор медицинских наук, профессор, руководитель группы по экспертизе МИ для in vitro диагностики департамента специализированной экспертизы медицинских изделий РГП на ПХВ «Национальный центр экспертизы лекарственных средств, изделий медицинского назначения и медицинской техники» МЗ РК.

— Год назад в Национальном центре экспертизы ЛС, ИМН и МТ РК при департаменте специализированной экспертизы МИ была создана группа по экспертизе МИ in vitro диагностики. Чем была вызвана необходимость создания отдельного подразделения для проведения экспертных работ по контролю качества медицинских изделий для диагностики in vitro?
Начнем с того, что основная задача медицины – лечить людей, а успешное лечение на 50 процентов зависит от правильной диагностики, которая, в свою очередь, невозможна без хороших и эффективных методов лабораторной диагностики.
Среди всего многообразия медицинских изделий, которые в настоящее время используются медиками во всех странах мира, большая их часть предназначена для диагностических целей. Это диагностика заболеваний и физиологических состояний (например, беременности), определение уровня различных показателей иммунной и эндокринной систем, биохимических показателей. То есть это целый арсенал методов, используемый врачами всех специализаций. Поэтому Всемирная организация здравоохранения разделила все МИ на две большие группы – для in vitro диагностики и не для in vitro диагностики.
В Казахстане в последние годы растет число лабораторий. Лаборатории есть в каждой клинике, поликлинике, научных центрах. Есть централизованные государственные лаборатории, обслуживающие государственные больницы и поликлиники. Появились частные, преимущественно сетевые, обслуживающие население Астаны, Алматы и других городов Казахстана. Для того чтобы оказываемая медицинская помощь населению страны была как можно более эффективной, все лаборатории должны использовать в своей работе качественные реагенты и лабораторную медицинскую технику. На рынок должны поступать только качественные МИ, отвечающие современным требованиям, созданные на основе последних научных достижений, поэтому необходима высококвалифицированная специализированная экспертиза МИ для in vitro диагностики при их регистрации в РК.
Все вышесказанное и явилось основанием для создания особой группы в Национальном центре экспертизы лекарственных средств, изделий медицинского назначения и медицинской техники. В состав группы вошли два профессора, доктора медицинских наук и один кандидат медицинских наук, имеющие большой опыт работы с МИ для in vitro диагностики, знающие особенности различных диагностических методов и применяемых диагностических реагентов и специального оборудования, прежде всего, различных анализаторов – биохимических, иммунологических, бактериологических и прочих. Для того чтобы экспертиза МИ для in vitro диагностики была качественной, на уровне мировых стандартов, нужно не только знать технологические особенности методов диагностики, но и уметь правильно оценить их значение при проведении клинических исследований, анализировать данные клинических испытаний, представляемые производителями. Если провести параллель с экспертизой лекарственных средств, то специалисты выполняют работу как по фармацевтической и фармакологической экспертизе, так и дают оценку клинических испытаний. Поэтому руководство нашего Центра в целях экспертизы МИ для in vitro диагностики сформировало отдельное подразделение.
— Как известно, единой методики контроля качества, которая подходит ко всем медицинским изделиям для диагностики in vitro, не существует. Какими нормативными документами регламентируется государственная экспертиза реагентов, инструментов, приборов, емкостей для сбора и хранения образцов или других изделий для in vitro диагностики?
Все НД, относящиеся к МИ для in vitro диагностики, перечислять не имеет смысла. Это и Приказы МЗ РК, и новые документы, разработанные рабочими группами ЕЭК.
Основа нормативной правовой базы – Кодекс РК «О здоровье народа и системе здравоохранения», приказы Министерства здравоохранения РК за номерами 736 и 735. Пользуемся мы также НПА, утвержденными Коллегией ЕЭК, действующими на территории всех пяти стран ЕАЭС. Положения, изложенные в указанных выше НПА, являются основой проведения экспертизы МИ, в том числе и МИ для in vitro диагностики.
Что касается универсальной методики контроля качества, то ее быть просто не может, так как лабораторных методов множество и они весьма специфичны. Например, метод контроля качества МИ, предназначенных для иммунной диагностики, не подходит для биохимической или генной диагностики.
Для каждого диагностического метода разрабатываются параметры определения эффективности, безопасности используемых реагентов и оборудования и их возможности применения в тех или иных медицинских исследованиях.
— Какими принципами (нормативными документами) должен руководствоваться производитель при разработке и изготовлении МИ для in vitro диагностики? Согласно международным стандартам и нормативным правовым актам, при разработке и изготовлении МИ ИВД производитель должен обратить особое внимание на оценку риска своего изделия в процессе его эксплуатации? С чем это связано?
Все производители МИ для in vitro диагностики должны быть аттестованы на соответствие международному стандарту СМК ИСО 13485. В ЕС основные правила, нормативы производства МИ изложены в специальной Директиве 98/79/ЕС. Положениями этой Директивы пользуются производители МИ практически всех стран мира.
Основной принцип производства МИ для in vitro диагностики формулируется очень просто: эффективность и безопасность. На словах это просто, на деле достаточно сложно. Что такое эффективность? МИ для in vitro диагностики, в первую очередь – высокая чувствительность и специфичность при выявлении определяемого аналита. Эти показатели исследуются уже на стадии разработки изделия. Применение данного МИ должно позволять выявлять искомый аналит как можно в более низкой дозе (концентрации), что будет определять его чувствительность, должно быть как можно меньше перекрестных реакций с другими веществами. Чем меньше возможности для перекрестных реакций, тем выше специфичность диагностики. Кроме того, показатель эффективности включает в себя удобство применения, стоимость изделия и прочие параметры.
Безопасность МИ для in vitro диагностики тоже понятие комплексное, и подходы к оценке их безопасности отличаются от подходов к оценке безопасности МИ, не предназначенных in vitro диагностики.
Для тех МИ, что вводятся в организм человека, понятие «безопасность» заключается в том, чтобы они не нанесли прямого вреда человеческому организму. При оценке безопасности МИ для in vitro диагностики оценивается как прямое неблагоприятное действие их на организм (химические вещества, входящие в состав наборов для диагностики, электромагнитное и другие виды излучений при использовании различных анализаторов), так и объект, с которым работают сотрудники лаборатории, используя тот ли иной диагностический реагент. Например, кровь. Существует около десятка известных сегодня возбудителей болезней, передающихся через кровь. Думаю, что неизвестных еще больше. По действующим НПД все МИ делятся на 4 класса безопасности. Согласно этой классификации реагенты, используемые для диагностики ВИЧ, гепатитов В и С, особо опасных инфекций, относятся к 3 (самому высокому) классу безопасности. Однако кровь служит исследуемым материалом не только при диагностике ВИЧ и гепатита, но и инфекциях, вызванных другими патогенными и условно-патогенными микроорганизмами. Реагенты, используемые для этого, уже будут относиться к низшим классам безопасности (2б или 2а). Получается, что опасность при исследовании одной и той же крови на ВИЧ и гепатиты и исследовании на другие инфекции, иммунный или гормональный статус, различается. На мой взгляд, это неправильный подход. Все реагенты, используемые для лабораторных анализов крови, должны иметь один класс безопасности. Ведь меры безопасности при проведении анализов на ВИЧ и кишечные инфекции должны быть одинаковыми, так как предметом исследования является кровь пациента. Врач отправляет больного на анализы, зачастую еще не зная, болен пациент или нет. Этот вопрос поднимается на всех уровнях. Недавно мы встречались с представителем ВОЗ, где я говорил о том, что, возможно, при классификации медицинских изделий для in vitro диагностики нужно уйти от классификации безопасности или дополнить их классификацию делением МИ, используемыми для иммунологической, генетической или биохимической диагностики. Сегодня мы смешали в одну кучу реагенты для сложных иммунологических и генетических исследований с реактивами для достаточно простых методов химического анализа. Если такое разделение будет внедрено, то гораздо проще будет относить такие методы к определенному классу, более правильно оценивать подходы в работе с ними. На мой взгляд, необходимо провести такую работу по разработке новой классификации медицинских изделий для in vitro диагностики.
Безопасность лабораторных исследований, в том числе крови, значительно повышается при использовании автоматизированных анализаторов, когда задача лабораторного работника – внести исследуемый материал, а все остальные процедуры машина выполняет без участия человека.
Таким образом, основным принципом, которым руководствуется разработчик и производитель, является создание высокочувствительных, высокоспецифичных и безопасных МИ для in vitro диагностики.
Однако, чем качественнее медицинское изделие, тем дороже его себестоимость. Особенно это касается МИ, предназначенных для новых инновационных методов диагностики. Например, в 80-90-е годы 20 века иммуноферментный анализ был экзотикой для нашей страны, реагенты стоили дорого. Сегодня это рутинный метод, используемый в лабораториях разного уровня – от крупных клиник до сельских амбулаторий. Массовое применение метода значительно снизило и стоимость реагентов. Наука не может стоять на месте, разрабатываются новые методы диагностики. И уже сегодня в Казахстане регистрируются МИ для различных видов генетической (полимеразная цепная реакция, генные и иммунные зонды, секвенирование и другие) и иммунологической (иммуноферментный анализ, иммунолюминесцентный анализ, иммунохемилюминисцентный анализ) диагностики. Регистрируется большое количество новых, самых современных автоматических анализаторов для биохимических, иммунологических, микробиологических, генетических исследований.
-Как (по какой методике или алгоритму) эксперты проверяют безопасность и качество медицинского изделия, хотя, как уже было сказано выше, единой методики контроля качества не существует? Чем отличается методика проверки качества и безопасности МИ ИВД в нашей стране от методик, применяемых в США, ЕС, бывших союзных республиках?
Алгоритм проведения экспертизы у всех один. Сначала проводится первичная экспертиза или, как сейчас называют, валидация регистрационного досье. Чем лучше и полнее будет проведена эта процедура, тем меньше времени будет потрачено при проведении специализированной экспертизы на формальные вопросы, например, определение НМИРК.
Следующий этап – аналитическая экспертиза, в ходе которой все МИ для in vitro диагностики должны пройти исследование в лаборатории. Это оценка основных показателей, прежде всего на соответствие заявленной чувствительности выявления аналита. Если в лабораториях нашего Испытательного центра нет возможности провести такие исследования (отсутствие необходимых микророганизмов, закрытый тип анализатора), то специалисты нашей группы совместно с сотрудником лаборатории выезжают на предприятие производителя для оценки условий производства и проведения аналитической экспертизы.
Затем очередь специализированной экспертизы, в ходе которой оценивается фактически эффективность и безопасность производства медицинского изделия и возможность его применения в условиях нашей страны.
Далее проводится оценка безопасности, затем документация передается Комитету фармации МЗ РК для принятия решения о выдаче регистрационного удостоверения.
Методики определения качества медицинских изделий in vitro диагностики во всех странах также едины. Если не будет единого подхода, то мы, эксперты, не сможем сравнивать. Поэтому все методы должны быть воспроизводимы.
Они не должны отличаться ни друг от друга, ни от метода производителя. Сейчас мы добиваемся унификации оценки качества МИ на всей территории ЕАЭС.
После регистрации медицинского изделия и его ввоза в страну контроль за его применением продолжается. Главным «контролером» является потребитель. Качество медицинских изделий, в том числе и изделий in vitro диагностики, контролируется посредством мониторинга их качества. Если потребитель выявил, что результаты исследований с использованием МИ не соответствуют по тем или иным показателям, указанным в инструкции по медицинскому применению, утвержденной в Казахстане, то он должен заполнить «желтую карту» и отправить ее в Национальный центр экспертизы ЛС, ИМН и МТ.
Подразделение Национального центра, занимающееся фармаконадзором и мониторингом качества МИ, должно на это отреагировать. Думаю, что пришло время создать отдельное подразделение и для мониторинга качества медицинских изделий, которых на рынке становится все больше.
Более того, считаю, что необходимо создать государственную структура, которая контролировала бы работу лабораторий, чтобы случаи, связанные с некачественным проведением анализов, тщательно анализировались с выявлением причин некачественной диагностики.
Это позволило бы также оценить подготовленность сотрудников лабораторий, от которых во многом зависит качество анализа и выбор метода исследования, оценить знания врачей клиницистов по использованию лабораторных методов диагностики, интепретации их результатов. К сожалению, я знаю много случаев, когда диагноз токсоплазмоза, например, верифицируется клиницистами по наличию IgG антител. Но это не показатель острой инфекции, а скорее последствия ранее перенесенных инфекций, свидетельство наличия иммунной защиты. А решение, например, для беременных женщин, может приниматься радикальное – аборт. Ситуация достаточно сложная в моральном аспекте, поэтому необходимо обучать не только лаборантов, но и клиницистов правильно интерпретировать результаты различных анализов, в том числе иммунологических.
-Для диагностики in vitro используются медицинские приборы открытого и закрытого типа. Расскажите подробнее о методиках испытаний этих МИ. Используется ли один подход или применяются разные подходы?
Термин «система закрытого типа» означает, что для работы этого анализатора необходимо использовать только определенные реагенты (расходные материалы), производимые чаще (но не всегда) производителем данного анализатора и поставляемые в комплекте с этим анализатором. Никакие другие реагенты использоваться не могут. О преимуществе систем закрытого типа я уже говорил ранее, их использование повышает безопасность, значительно сокращается контакт лабораторных сотрудников с исследуемым материалом. Обычно это инновационная техника с высокой чувствительностью и производительностью. Недостаток их связан с тем, что пользователь привязан к расходным материалам (реагентам) только одного производителя. Отсюда отсутствие конкуренции и, чаще всего, высокая стоимость расходных материалов. В тех случаях, когда используется анализатор открытого типа, можно применять реагенты разных производителей. Такой анализатор удобен тем, что нет зависимости от одного производителя. Но с другой стороны, анализаторы закрытого типа часто обладают уникальными возможностями, широким спектром выявляемых аналитов, большей производительностью, дают информацию для расширенного анализа. Только в процессе работы отбирается то, что нужно для проведения качественного анализа.
— Этот вопрос хочу задать, как пациент. В последнее время многие казахстанцы начали сомневаться в результатах анализов. Говорят, что в разных лабораториях они могут очень отличаться. Врачи советуют сдавать анализы в нескольких лабораториях для установления более точного диагноза, на что уходит немало времени и средств. Отчего сложилась такая ситуация, ведь в лабораториях РК (частных и государственных) используют одни и те же реагенты и медицинские изделия, прошедшие государственную регистрацию?
Как я уже говорил, лабораторий во всех городах Казахстана становится все больше и больше, что требует самых действенных мер для их контроля.
Не могу утверждать, что сегодня все лаборатории дают абсолютно правильные результаты. Мы, эксперты, свою задачу выполняем, рекомендуя только МИ с доказанной эффективностью и безопасностью. Но для того чтобы их применять, необходимы качественная аппаратура и знания. Надо не только контролировать, но и обучать, причем не разово, а на системной основе. Методы всегда совершенствуются: обучим одному, уже на смену пришло что-то более совершенное.
Были случаи, когда результаты одних и тех исследований в разных лабораторий сильно различаются. Например, по лабораторной верификации вирусного гепатита В: одна лаборатория выдает один результат, другая другой. Ни пациенту, ни врачу ничего не ясно. Поэтому в Казахстане создана референс лаборатория диагностики вирусных гепатитов в Научно—практическом центре санитарно-эпидемиологической экспертизы и мониторинга (бывшая Республиканская СЭС). Если у больного или врача возникли сомнения по поводу полученных результатов, они могут повторить анализы в этой лаборатории.
А обучать наших специалистов лабораторий надо обязательно на системной основе. Оборудование в лабораториях большей частью довольно сложное, дорогостоящее, реагенты закупаются у разных производителей, поэтому необходимо уметь правильно использовать современные методики проведения анализов.
—Вопрос, вытекающий из предыдущего: каким испытаниям подвергаются МИ ИВД, прежде чем будут взяты на вооружение в лабораториях нашей страны?
Любой разработчик, прежде всего, сам испытывает свою продукцию. Медицинские изделия для in vitro диагностики проходят ряд обязательных испытаний. Например, оценивается лабораторная чувствительность выявления аналита, специфичность выявления (не дает ли реакции с какими-то другими аналитами). Такого рода испытания проводятся иногда на лабораторных животных, иногда на панели каких-либо сывороток, антигенов.
Еще один важный показатель качества МИ (реагентов, реактивов) – стабильность. Определяется срок его хранения, то есть время, в течение которого они при хранении в определенных условиях сохранят свои свойства, прежде всего, чувствительность и специфичность. Сегодня все имеющиеся на рынке реагенты и реактивы для in vitro диагностики имеют достаточно длительные сроки хранения: некоторые – 6-9 месяцев, но обычно – 1-3 года. Это обстоятельство позволяет транспортировать такие реагенты практически из любой точки мира и создавать необходимые для лабораторий запасы.
Затем обязательно проводятся клинические (медицинские) испытания данного МИ параллельно с принятыми ранее методами для выявления этих же показателей. Клинические испытания могут проводиться как в стране производителя, так и в других странах. Только после проведения всех лабораторных и клинических испытаний новые медицинские изделия попадают к нам на экспертизу. Наше заключение после экспертизы входит в перечень обязательных документов.
-Вы, Павел Николаевич, всю жизнь посвятили изучению и профилактике наиболее опасных для человечества инфекционных болезней. Только в этом году в СМИ была озвучена информация о случаях холеры в Алматы, обнаружении инфекции непонятной этиологии в одном из детских садов Актобе. Также инфекционные заболевания периодически вызывают падеж скота в том или ином регионе нашей страны. Люди пугают друг друга «страшилками» о зараженном мясе на базарах, молочных продуктах импортного производства с кишечными палочками. Проводится ли in vitro диагностика штаммов особо опасных инфекций в Казахстане? Закупается ли для этого медицинское оборудование и реагенты, и можно ли доверять результатам исследований в этом направлении?
Да, я всю жизнь посвятил инфектологии, в частности, лабораторной диагностике и профилактике инфекционных заболеваний, в том числе особо опасных. Информация о случаях холеры, насколько я знаю, не была подтверждена, это были кишечные инфекции, вызванные другими возбудителями. Неясные случаи заболеваний инфекционного (а может и не инфекционного) характера, как это было в Актобе, к сожалению, встречаются в педиатрической практике. Для постановки более точного диагноза необходимо проводить тщательные лабораторные исследования с большим набором реагентов бактериальной и вирусной специфичности. Необходимо провести тщательное эпидемиологическое расследование этих случаев для установления источника заражения.
К вопросу о продуктах. Если вы будете покупать мясо вне официальных рынков, где не осуществляется ветеринарный контроль, то сильно рискуете здоровьем, ведь уровень заболеваемости бруцеллезом в нашей стране достаточно высок. Там Вы можете даже купить мясо, зараженное сибирской язвой. Поэтому нужно обезопасить себя и свою семью, тщательно выбирая место для покупки мясных и молочных продуктов. Также важно правильно проводить их термическую обработку, прежде чем употреблять в пищу. При правильной термической обработке погибает практически 100% возбудителей, особенно если это бруцеллез. Нельзя покупать молоко на стихийных рынок или у торговок вдоль автомобильных трасс, так как в этом случае нет уверенности, что корова или коза не болеют бруцеллезом, а лошадь не пала от сибирской язвы. Поэтому лучше покупать там, где есть ветеринарный контроль.
Всеми проблемами особо опасных инфекций, включая выделение и исследование штаммов возбудителей, занимаются специалисты МЗ РК. В первую очередь это сотрудники Научного центра карантинных и зоонозных инфекциями имени М. Айкимбаева. На территории этого научного центра недавно построена и введена в строй так называемая Центральная референс лаборатория, которую построили в Алматы американцы. Ее основная цель – диагностика особо опасных инфекций, здесь также хранятся все штаммы возбудителей особо опасных инфекций, которые до сих пор хранились в Научном центре карантинных и зоонозных инфекций (чумы, сибирской язвы и других бактериальных возбудителей). Центр оснащен самым современным оборудованием, позволяющим выполнять любые лабораторные исследования нужного профиля. Я думаю, что для определения особо опасных инфекций в стране закупается нужное оборудование и реагенты. Я неоднократно говорил, что единственный, но весьма существенный минус – отсутствие производства собственных вакцин от особо опасных инфекций, за исключением чумы.
— Изменился ли качественно процесс экспертизы и регистрации МИ ИВД после создания Вашей группы? Что было сделано для более качественной экспертизы такого рода оборудования за прошедший год?
Конечно, качественно существенно изменился, стал более полным и глубоким благодаря профессионализму профессора Ж.А. Сатыбалдиевой, доцента Е.В. Хайрушева и вашего покорного слуги. Мы – специалисты в области лабораторной диагностики. Будь то инфекции, аллергические заболевания или оценка иммунного и гормонального статусов. И наши знания, накопленные годами практики, способствуют качественному проведению экспертизы регистрационных досье на МИ для in vitro диагностики. На мой взгляд, наша высокопрофессиональная группа в разы улучшила качество государственной экспертизы медицинских изделий, используемых для проведения in vitro диагностики.
Бактерии с «необычной» антибиотикорезистентностью широко распространены в США
 Центры по контролю и профилактике заболеваний США (Centers for Disease Control and Prevention – CDC) сообщают, что в прошлом году в США было выявлено более 220 бактерий с «необычными» генами устойчивости к антибиотикам.
Центры по контролю и профилактике заболеваний США (Centers for Disease Control and Prevention – CDC) сообщают, что в прошлом году в США было выявлено более 220 бактерий с «необычными» генами устойчивости к антибиотикам.
Они включают бактерии, которые:
- устойчивы к большинству (или всем) известным антибиотикам;
- распространены на нехарактерной для них географической территории;
- имеют специфические гены, которые позволяют им «делиться» антибиотикорезистентностью с другими бактериями.
Быстрое выявление новых или редких инфекционных угроз является важнейшим первым шагом в стратегии сдерживания распространения антибиотикорезистентности в США. Данная стратегия требует быстрой идентификации резистентности у микроорганизмов, оценки инфекционного контроля, тестирования пациентов без симптомов, которые могут переносить и распространять опасные бактерии, и продолжение оценки инфекционного контроля до прекращения распространения возбудителя. Это реализуется с помощью скоординированных ответных мер между медицинскими учреждениями, лабораториями, департаментами здравоохранения и CDC через сеть AR Lab.
Новые данные свидетельствуют о том, что стратегия сдерживания распространения антибиотикорезистентности в США может предотвратить тысячи трудно подающихся лечению или потенциально неизлечимых инфекций, включая высокоприоритетные угрозы, — таких возбудителей, как Candida auris и устойчивых к карбапенему штаммов энтеробактерий.
По оценкам, благодаря стратегии сдерживания распространения антибиотикорезистентности в США за 3 года было предотвращено до 1600 новых случаев инфекций, вызванных устойчивыми к карбапенему штаммами энтеробактерий только в одном штате.
Микроорганизмы будут постоянно находить способы противостоять новым и существующим антибиотикам. Остановить этот процесс невозможно, однако его можно сдерживать и контролировать, чтобы не допустить распространения опасных устойчивых к антибиотикам штаммов бактерий в медицинских учреждениях.
cdc.gov
В США впервые за последние 40 лет одобрен препарат для лечения лимфомы ходжкина на поздних стадиях
 Управлением по контролю за пищевыми продуктами и лекарственными средствами США (U.S. Food and Drug Administration – FDA) одобрило Adcetris (брентуксимаб ведотин) для лечения взрослых пациентов с классической лимфомой Ходжкина III или IV стадии, ранее не проходивших лечение, в сочетании с химиотерапией.
Управлением по контролю за пищевыми продуктами и лекарственными средствами США (U.S. Food and Drug Administration – FDA) одобрило Adcetris (брентуксимаб ведотин) для лечения взрослых пациентов с классической лимфомой Ходжкина III или IV стадии, ранее не проходивших лечение, в сочетании с химиотерапией.
Данный препарат обеспечит улучшение начальных схем лечения лимфомы Ходжкина на продвинутых стадиях, которые были введены в клиническую практику более 40 лет назад. Adcetris также был ранее одобрен FDA для лечения лимфомы Ходжкина после рецидива либо после трансплантации стволовых клеток, когда пациент подвергается высокому риску рецидива или прогрессирования; системной анапластической крупноклеточной лимфомы и первичной кожной анапластической крупноклеточной лимфомы в случаях неудачи предыдущего лечения.
Adcetris объединяет антитело и лекарственное средство, позволяя антителу направлять действующее вещество на мишень – клетки лимфомы, известные как CD30. Основанием для одобрения препарата стало клиническое исследование при участии взрослых пациентов с классической лимфомой Ходжкина III или IV стадии, ранее не проходивших лечение. В рамках исследования сравнивалась эффективность лечения Adcetris в сочетании с химиотерапией (с режимом лечения только химиотерапией). В ходе исследования была измерена модифицированная выживаемость пациентов без прогрессирования, которая учитывает продолжительность периода времени до прогрессирования болезни, смерти или необходимости в проведении новой терапии вследствие отсутствия полного ответа на проведенное лечение.
В исследовании при участии 1 334 пациента после того, как они получали в среднем шесть 28-дневных курсов лечения, у пациентов, получавших Adcetris в сочетании с химиотерапией, была отмечена на 23% меньшая вероятность прогрессирования заболевания, смерти или необходимости начала новой терапии по сравнению с пациентами, получающими только химиотерапию. В группе, получавшей Adcetris в сочетании с химиотерапией, было 117 (18%) пациентов, у которых было отмечено прогрессирование заболевания, смерть или необходимость в начале новой терапии по сравнению с 146 (22%) пациентами из группы, получавшей только химиотерапию.
Общие побочные эффекты Adcetris включают нейтропению, анемию, периферическую невропатию, тошноту, усталость, запор, диарею, рвоту и лихорадку. В вышеуказанном клиническом исследовании у 67% пациентов, получавших Adcetris в сочетании с химиотерапией, наблюдалась периферическая невропатия, у 91% – нейтропения. Профилактическое лечение G-CSF, фактором роста костного мозга для производства белых кровяных телец, рекомендуется при лечении Adcetris в сочетании с химиотерапией в рамках первого курса лечения лимфомы Ходжкина III или IV стадии.
При применении Adcetris существует риск заражения вирусом Джона Каннингема, который может привести к прогрессивной мультифокальной лейкоэнцефалопатии, редкой, но тяжелой инфекции головного мозга, вследствие которой может наступить смерть.
Тяжелые побочные эффекты при применении Adcetris включают периферическую невропатию, тяжелые аллергические реакции (анафилаксия) или реакции на введение препарата, гематологическую, легочную и гепатотоксичность, тяжелые или оппортунистические инфекции, метаболические нарушения (синдром лизиса опухоли), тяжелые дерматологические реакции и осложнения со стороны ЖКТ. Adcetris может нанести вред развивающемуся плоду и ребенку, находящемуся на грудном вскармливании.
fda.gov
ОГРАНИЧЕНИЯ ОТНОСИТЕЛЬНО ПРИЕМА ВАЛЬПРОАТОВ ДЛЯ ЖЕНЩИН
 Координационная группа по взаимному признанию и децентрализованным процедурам (Group for Mutual Recognition and Decentralised Procedures – CMDh) одобрила новые меры, чтобы избежать влияния вальпроатов на внутриутробное развитие детей, поскольку у детей, подвергшихся такому влиянию во время беременности матери, повышается риск возникновения пороков развития.
Координационная группа по взаимному признанию и децентрализованным процедурам (Group for Mutual Recognition and Decentralised Procedures – CMDh) одобрила новые меры, чтобы избежать влияния вальпроатов на внутриутробное развитие детей, поскольку у детей, подвергшихся такому влиянию во время беременности матери, повышается риск возникновения пороков развития.
Таким образом, одобренные CMDh меры усиливают прежние ограничения относительно использования вальпроатов и требования к информированию о риске, связанном с их приемом для женщин.
Вальпроат содержащие лекарства были одобрены на национальном уровне в ЕС для лечения эпилепсии и биполярного расстройства, а в некоторых странах для профилактики мигрени. Новые меры включают запрет на применение этих лекарственных средств для лечения мигрени или биполярного расстройства во время беременности, а также запрет на лечение ими эпилепсии во время беременности, за исключением случаев, когда нет другого эффективного варианта терапии.
Кроме того, вальпроатсодержащие препараты не должны применяться у представительниц женского пола, способных к зачатию ребенка, при несоблюдении мер по предотвращению беременности в рамках новой программы. Эта программа предназначена для того, чтобы пациенты полностью осознавали риски и необходимость избегать беременности во время приема определенных лекарств.
Визуальное предупреждение о рисках, связанных с приемом вальпроатов во время беременности, также должно быть размещено на упаковке вальпроат содержащих препаратов.
Компании, продающие эти лекарства, должны проводить дополнительные исследования относительно характера и масштабов влияния рисков, связанных с приемом вальпроатов, контролировать применение этих лекарств и долгосрочные последствия их влияния на детей, чьи матери применяли вальпроаты во время беременности.
ema.europa.eu
ОБЕЗБОЛИВАЮЩЕЕ ФЛУПИРТИН БУДЕТ ОТОЗВАНО С РЫНКА ЕС
Координационная группа по взаимному признанию и децентрализованным процедурам (Group for Mutual Recognition and Decentralised Procedures – CMDh) одобрила рекомендацию EMA отозвать лицензию на маркетинг лекарств на основе флупиртина из-за риска серьезного поражения печени. Это означает, что препарат больше не будет доступным на рынке ЕС.
Рекомендация EMA была результатом обзора лекарственных средств на основе флупиртина, который был начат ввиду того, что сообщения о проблемах с печенью продолжали поступать даже после того, как в 2013 г. были введены меры по управлению этим риском при приеме данных лекарств. Это ограничение применения флупиртина сроком не более чем 2 недели у пациентов с острой болью, которые не могли принимать другие обезболивающие средства, а также проведение еженедельных тестов для оценки функции печени во время лечения флупиртином.
В обзоре, проведенном Комитетом по оценке риска фармаконадзора (Pharmacovigilance Risk Assessment Committee – PRAC), были рассмотрены все имеющиеся данные, включая исследования, оценивающие соблюдение ограничительных мер при применении флупиртина, введенных в 2013 г. в клиническую практику. Также оценивались случаи серьезного поражения печени, которые были зарегистрированы после обзора 2013 г.
CMDh согласилась с заключением PRAC о том, что ограничения, введенные в 2013 г., соблюдались в недостаточной мере, и случаи серьезного повреждения печени, включая печеночную недостаточность, при применении флупиртина все еще имели место. Кроме того, не было предпринято никаких дальнейших мер по усилению соблюдения введенных ограничений и адекватному снижению риска развития проблем с печенью при приеме этого обезболивающего.
Поэтому CMDh согласилась с тем, что пациенты, принимающие лекарства, содержащие флупиртин, продолжают подвергаться серьезным рискам, которые перевешивают выгоды этих лекарств. В целях защиты общественного здоровья CMDh одобрила рекомендацию PRAC относительно отзыва лицензий на маркетирование лекарств, содержащих флупиртин.
ema.europa.e
Сердечно-сосудистая безопасность кларитромицина US Food and  Drug Administration (FDA) настаивает на изучении сердечно-сосудистой безопасности кларитромицина у пациентов с ишемической болезнью сердца (ИБС).
Drug Administration (FDA) настаивает на изучении сердечно-сосудистой безопасности кларитромицина у пациентов с ишемической болезнью сердца (ИБС).
Согласно результатам и10-летнего проспективного плацебо-контролируемого исследования CLARICOR у пациентов с ИБС отмечается повышение смертности после 2 недельного курса кларитромицина. На основании этих данных FDA добавило новое предупреждение в инструкцию к препарату и советует выбирать альтернативный антибиотик у больных ИБС.
Кларитрмицин широко используется в лечении инфекций кожи, ушей, синусов, легких, инфекции H. pylori, а также инфекций Mycobacterium avium, и поражений легких у ВИЧ-инфицированных лиц.
- Эксперты FDA отмечают, что на данный момент четкого понимания каким образом кларитромицин повышает смертность нет. Обращает на себя внимание, что в дополнение к исследованию CLARICOR в 2 из 6 обсервационных исследований показаны долгосрочные неблагоприятные риски кларитромицина.
Вопрос требует пристального внимания, в настоящее время FDA продолжает анализировать сообщения о нежелательных явлениях со стороны кларитромицина.
Источник: FDA review finds additional data supports the potential for increased long-term risks with antibiotic clarithromycin (Biaxin) in patients with heart disease. February 2018.
Annals of Internal Medicine 23 January 2018.
(internist.ru)
БАКТЕРИИ ПРИОБРЕТАЮТ УСТОЙЧИВОСТЬ ДАЖЕ К СПИРТУ
 Резистентные бактерии продолжают приобретать новые способности. Последние исследования показали, что теперь они научились приспосабливаться к санитайзерам и комплексным антисептикам, которые использует больничный персонал для обработки рук.
Резистентные бактерии продолжают приобретать новые способности. Последние исследования показали, что теперь они научились приспосабливаться к санитайзерам и комплексным антисептикам, которые использует больничный персонал для обработки рук.
Как выяснили ученые из Мельбурнского университета, в течение последних 20 лет микроорганизмы стали в 10 раз устойчивее к спирту, который входит в состав таких антисептиков.
Одни те же антисептические средства для обработки рук используются в больницах уже несколько десятилетий, поэтому их состав в настоящее время уже устарел. Раньше специалисты полагали, что специальные жидкости могут остановить распространение опасных микробов, особенно ванкомицин-резистентных энтерококк (vancomycin resistant Enterococcus faecium, VRE Efm) и метициллин-резистентный золотистый стафилококк (Staphylococcus aureus).
Супербактерии – это микроорганизмы, которые смогли выработать устойчивость к антимикробным препаратам. В последнее два десятилетия они стали одним из главных поводов для беспокойства медицинского сообщества. Проблема антимикробной резистентности (АМР) становится глобальной. Микробы, как и все живые существа, адаптируются к меняющейся среде. Из-за этого эффективность существующих антибиотиков исчерпывается, а разработка новых занимает много времени и стоит очень дорого.
По данным ВОЗ, в числе самых распространенных резистентных бактерий значатся:
- Метициллин-резистентный золотистый стафилококк (Staphylococcus aureus, MRSA)
- Ванкомицин-резистентный энтерококк (Enterococcus faecium)
- Кишечная палочка (Escherichia coli), резистентная к цефалоспоринам 3-го поколения
- Клебсиелла пневмония (Klebsiella pneumoniae), резистентная к цефалоспоринам 3-го поколения
- Синегнойная палочка (Pseudomonas aeruginosa), резистентная к карбапенемам
Несмотря на то, что эти микроорганизмы развили устойчивость к антибиотикам, ученые полагали, что они все же не смогут приспособиться к спирту, так как он мгновенно повреждает внешние мембраны микробов.
Команда ученых из Мельбурнского университета под руководством профессора Тимоти СТАЙНЕРА (Timothy Stinear) протестировала на устойчивость к спирту 139 видов больничных штаммов Enterococcus faecium, взятых в период с 1997 по 2015 годы. Оказалось, что штаммы бактерий, отобранных для эксперимента после 2010 года, в 10 раз устойчивее к спиртовым растворам, чем их более ранние образцы. К тому же ученые выяснили, что «современные» штаммы Enterococcus faecium успешно сопротивлялись даже 70-процентному раствору изопропилового спирта.
Эксперты предположили, что подобное произошло из-за накопления мутаций, модифицирующих гены Enterococcus faecium таким образом, что изопропиловый спирт, как и другие спирты, стал принимать участие в собственном метаболизме бактерий.
Ученые считают, что необходимо срочно выработать другие методы антисептической обработки рук в больницах, так как Enterococcus faecium является чрезвычайно опасной инфекцией, устойчивой к антибиотикам последнего поколения, и, по меньшей мере, половина пациентов, инфицированных такими резистентными штаммами, погибает.
Юлия БОНДАРЬ,
mednovosti.ru
ПРЕПАРАТЫ ВАЛЬПРОАТА В ФОКУСЕ БЕЗОПАСНОСТИ ВО ВРЕМЯ БЕРЕМЕННОСТИ
 Запрет на использование во время беременности препаратов, содержащих вальпроат, является одной из самых обсуждаемых тем. Данные реальной клинической практики во Франции свидетельствуют о том, что в 30-40% случаев у детей, матери которых принимали вальпроат во время беременности, имеется риск серьезных нарушений развития и поведения, а в 10% случаев имеется риск тяжелых врожденных мальформаций.
Запрет на использование во время беременности препаратов, содержащих вальпроат, является одной из самых обсуждаемых тем. Данные реальной клинической практики во Франции свидетельствуют о том, что в 30-40% случаев у детей, матери которых принимали вальпроат во время беременности, имеется риск серьезных нарушений развития и поведения, а в 10% случаев имеется риск тяжелых врожденных мальформаций.
Вальпроат активно и повсеместно используется для лечения эпилепсии, биполярных расстройств и иногда профилактики мигрени.
Фармакологический надзор Европейского медицинского агентства (PRAC) изучил имеющиеся данные по применению вальпроата во время беременности. Учитывая высокий риск отрицательного воздействия на плод, PRAC выпустил рекомендации.
Для лечения женщин детородного возраста с мигренью и биполярными расстройствами и беременных женщин вальпроат не должен быть использован.
У беременных женщин с эпилепсией следует избегать применение вальпроата. В случае, когда прекратить терапию вальпроатом не представляется возможным, необходимо более тщательное наблюдение во время беременности. У женщин детородного возраста с эпилепсией следует избегать назначения вальпроата. В случае принятия решения о применении вальпроата необходимо проведение теста на беременность до и во время терапии.
PRAC считает необходимым изменение упаковки препаратов, содержащих вальпроат. По мнению экспертов, важно визуальное предупреждение о рисках во время беременности, например, при помощи пиктограмм, символов, адаптированных для каждой страны.
Источник:
PRAC recommends new measures to avoid valproate exposure
in pregnancy,
9 February 2018
(internist.ru)
ПРИМЕНЕНИЕ СТЕРОИДОВ ПРИ ХОБЛ УВЕЛИЧИВАЕТ РИСК ПЕРЕЛОМОВ ПЛЕЧА И ШЕЙКИ БЕДРА
Хроническая обструктивная болезнь легких (ХОБЛ) вызывается непрекращающимся воспалением бронхов и их прогрессирующим сужением. По статистике, в течение жизни ХОБЛ развивается у каждого второго курильщика. Чтобы уменьшить воспаление на поздних стадиях, таким пациентам часто назначаются ингаляционные глюкокортикостероиды. В то же время эти препараты обладают большим количеством нежелательных эффектов, и безопасность такой терапии остается под вопросом.
Безопасность назначения ГКС при ХОБЛ оценили специалисты Центра здоровья университета МакГилла в Монреале (McGill University Health Centre, Montreal). Они анализировали связь между длительным использованием ингаляционных глюкокортикостероидов при ХОБЛ и риском переломов плеча или шейки бедра. Результаты своего исследования авторы опубликовали в последнем номере журнала Chest.
Авторы проанализировали данные 240 тысяч пациентов с ХОБЛ, средний период наблюдения за которыми составил 5 лет. За это время произошло 19 тысяч переломов плеча или шейки бедра. При этом значительно чаще переломы происходили у пациентов, принимавших глюкокортикостероиды. Использование ингаляционных ГКС в дозировке, по крайней мере, 1 мкг на протяжении 4 лет приводило к значительному увеличению риска (относительный риск 1,1; 95% доверительный интервал 1,02-1,19).
Ранее аналогичная зависимость была установлена и для пациентов с бронхиальной астмой, которые тоже пользуются стероидными препаратами. По мнению канадских специалистов, каждый случай назначения глюкокортикостероидов требует тщательной оценки риска и пользы потенциальной терапии. Зачастую назначение этого класса препаратов, особенно на ранних стадиях заболевания, неоправданно, и можно обойтись более безопасными средствами. За людьми, длительно использующими гормональную терапию при ХОБЛ, авторы призывают наблюдать особо.
mednovosti.ru
ОПАСНОСТЬ ПРИМЕНЕНИЯ ТИРЕОСТАТИЧЕСКИХ ПРЕПАРАТОВ ВО ВРЕМЯ БЕРЕМЕННОСТИ
 Согласно результатам нового исследования, представленного в Annals of Internal Medicine, применение антитиреоидных препаратов в первом триместре беременности, ассоциировано с повышением риска врожденных пороков развития.
Согласно результатам нового исследования, представленного в Annals of Internal Medicine, применение антитиреоидных препаратов в первом триместре беременности, ассоциировано с повышением риска врожденных пороков развития.
Дизайн исследования
Исследователи из Сеула (Корея) проанализировали данные национального когортного исследования на предмет ассоциации между применением антитиреоидных препаратов и мальформаций у детей.
Когорта включала 2 886 970 беременностей, закончившихся рождением живых детей у 2 210 253 женщин (2008-2010 гг.) За период наблюдения был выявлен 12 891 случай применения тиреостатических препаратов на протяжении первого триместра гестации (0,45%).
Результаты
Распространенность врожденных пороков развития у детей, чьи матери получали тиреостатические препараты, составила 7,27% по сравнению с 5,94% в контрольной группе. Скорректированное отношение шансов, 1,19 95% CI, 1,12-1,28 (P < 0,001).
Абсолютное повышение частоты врожденных пороков на 1000 живых детей составило 8,81 случая для пропилтиоурацила (95% CI, 3,92-13,70 случаев), 17,05 случаев (95% CI, 1,94-32,15 случаев) для метимазола и 16,53 случая (95% CI, 4,73-28,32 случаев) для комбинации пропилтиоурацила и метимазола по сравнению с контрольной группой.
В группе метимазолавысокая кумулятивная доза (>495 мг), используемая в 1 триместре, была ассоциирована с более высоким риском врожденных пороков развития по сравнению с дозой 1-126 мг (отношение шансов 1,87 [95% CI, 1,06-3,30].
Заключение
Применение тиреостатических препаратов в период первого триместра беременности сопряжено с достоверным повышением риска врожденных пороков развития.
Источник:
Gi Hyeon Seo, Tae Hyuk Kim, Jae Hoon Chung.
В ЕС БУДЕТ ПРИОСТАНОВЛЕН ОБОРОТ РАСТВОРОВ ДЛЯ ИНФУЗИЙ, СОДЕРЖАЩИХ ГИДРОКСИЭТИЛКРАХМАЛ
 Координационная группа по взаимному признанию и децентрализованным процедурам (Group for Mutual Recognition and Decentralised Procedures – CMDh) одобрила рекомендацию о приостановлении маркетирования растворов для инфузий, содержащих гидроксиэтилкрахмал (ГЭК) в ЕС.
Координационная группа по взаимному признанию и децентрализованным процедурам (Group for Mutual Recognition and Decentralised Procedures – CMDh) одобрила рекомендацию о приостановлении маркетирования растворов для инфузий, содержащих гидроксиэтилкрахмал (ГЭК) в ЕС.
Препараты, содержащие ГЭК, используются для лечения гиповолемии (низкий объем крови), вызванной острой (внезапной) потерей крови, когда лечение альтернативными инфузионными растворами, известными как кристаллоидные, считается недостаточным.
Приостановка оборота препаратов, содержащих ГЭК, связана с тем, что эти лекарства продолжают использоваться у пациентов в критическом состоянии, пациентов с сепсисом и нарушением функции почек, несмотря на ограничения относительно применения таких препаратов, введенные в 2013 г. с целью снижения риска возникновения проблем с почками и случаев смерти пациентов.
Обзор данных относительно препаратов, содержащих ГЭК, был проведен Комитетом по оценке риска фармаконадзора (Pharmacovigilance Risk Assessment Committee – PRAC) Европейского агентства по лекарственным средствам (European Medicines Agency – EMA), который проанализировал результаты исследований относительно применения этих препаратов, имеющиеся данные об их пользе/риске в рамках клинических, обсервационных исследований, а также отзывы, полученные от заинтересованных сторон и экспертов. На основе этого обзора PRAC пришел к выводу, что ограничения, введенные в 2013 г., недостаточно эффективны. PRAC также изучил возможность введения дополнительных мер для защиты пациентов, однако считает, что такие меры будут неэффективными или недостаточными. В связи с этим CMDh согласилась с рекомендацией PRAC, что с учетом серьезных рисков, которым подвергаются определенные пациенты, оборот препаратов, содержащих ГЭК, должен быть приостановлен. В настоящее время доступны альтернативные варианты лечения.
ema.europa.eu
ПРАВИЛА ПЕРЕХОДА С ОДНОГО ПРОТИВОЭПИЛЕПТИЧЕСКОГО ПРЕПАРАТА НА ДРУГОЙ
 Актуальность
Актуальность
Медицинское агентство Великобритании провело реклассификацию противоэпилептических препаратов, выделив 3 категории лекарств. Это было сделано по просьбе пациентов и специалистов здравоохранения.
Известно, что переход с одного торгового бренда на другой может сопровождаться повышением частоты побочных эффектов иухудшением течения заболевания. Разделение препаратов на 3 категории позволяет определить правила смены препаратов.
3 категории противоэпилептических препаратов
К препаратам первой категории относятся карбамазепин, фенобарбитал, фенитоин ипримидон. У данных препаратов выявлены клинически значимые различия между различными брендами. Отмечается, что различия могут возникнуть даже между биоэквивалентыми препаратами. В этой связи целесообразно назначать один и тот же бренд.
Препаратами второй категории являются клоназепам, эсликарбазепин, ламотриджин,оксарбазепин, перампанел, топирамат, вальпроат, зонисамид. Это лекарственные средства, которые не попадают под категорию 1 и 3. При выписке препарата целесообразно придерживаться того же торгового бренда.
К лекарственным средствам третьей категории отнесены бриварацетам, этосуксимид, габапентин, лакосамид, леветирацетам, прегабалин, тиагабин и вигабатрин.
Данные препараты имеют следующие характеристики: высокую растворимость в зависимости от рН, высокую абсорбцию при пероральном приеме, не выраженную дозозависимую эффективность и безопасность, узкий терапевтический диапазон.
Необходимо отметить, что клинически значимые различия между различными брендами встречаются крайне редко. При решении вопроса о смене бренда важно оценить частоту эпилептических кризов и лекарственный анамнез пациента.
internist.ru
(Источник:
Drug Safety Update volume 11, issue 4;November 2017: 5)
FDA ВВОДИТ ОГРАНИЧЕНИЕ НА ОБЪЕМ УПАКОВКИ ЛОПЕРАМИДА

Food and Drug Administration (FDA) вводит ограничения на объем упаковки противодиарейного препарата лоперамид. Это делается с целью повышения безопасности терапии.
В последнее время отмечается учащение злоупотребления лоперамидом и увеличение случаев сердечно-сосудистых побочных эффектов (обморок, удлинение интервала QT, нарушение ритма по типу «torsades de pointes» или других желудочковых аритмий, остановка сердца).
Максимально разрешенной дозой лоперамида для взрослых считается 8 мг при безрецептурном применении и 16 мг в день при назначении врачом. В разрешенных дозах препарат считается безопасным, однако превышение дозы чревато тяжелыми последствиями – серьезной аритмией и смертью.
Большинство случаев злоупотребления лоперамидом описываются у пациентов, принимающих одновременно другие препараты, повышающие его всасывание, а также подавляющие метаболизм лоперамида, что приводит к развитию эйфорического эффекта. Помимо этого, некоторые лица принимают лоперамид для лечения симптомов отмены опиоидов.
Важно донести до пациентов информацию, что в случае продолжения диареи на фоне лоперамида на протяжении более 2 дней нужно обратиться за медицинской помощью. Помимо этого, потеря сознания, учащенное сердцебиение или нерегулярный сердечный ритм, спутанность сознания требуют срочного обращения за неотложной помощью.
Источник:
FDA Drug Safety Communication:
FDA limits packaging for anti-diarrhea medicine Loperamide (Imodium) to encourage safe use. Drug Safety and Availability. January 2018.
internist.ru
НЕБЕЗОПАСНЫЕ АНТИБИОТИКИ ВО ВРЕМЯ БЕРЕМЕННОСТИ ПРОДОЛЖАЮТ НАЗНАЧАТЬ

Как показали результаты нового исследования, сульфониламидные антибиотики и нитрофурантоин, не смотря на противопоказания, часто назначаются в первом триместре при инфекциях мочевых путей. При этом применение данных препаратов повышает риск врожденных пороков развития.
Исследователи из Центра по контролю и профилактике заболеваний США (CDC) проанализировали назначения антибактериальных препаратов у беременных в 2013-2015 годах. Было идентифицировано 482 917 беременностей у женщин в возрасте от 15 до 44 лет.
У 41% женщин диагностированы внебольничные инфекции мочевых путей в первом триместре, тогда как на протяжении 90 дней до беременности – у 7,2%.
Результаты
Среди женщин с диагностированными мочевыми инфекциями 68,9% получали антибиотик, если речь шла об инфекции во время 1 триместра, по сравнению с 76,1% женщин в период до беременности.
Не беременным женщинам чаще назначали фторхинолоны и сульфониламиды. А наиболее часто назначаемыми препаратами для лечения мочевых инфекций в первом триместре являлись нитрофурантоин (34,7%), ципрофлоксацин (10,5%), цефалексин (10,3%) и триметоприм-сульфаметоксазол (7,6%).
Обращает на себя внимание, что 4 из 10 женщин получали антибактериальную терапию, не рекомендованную во время беременности.
Авторы публикации подчеркивают, что врачам следует обращать внимание на риски, ассоциированные с назначением антибиотиков на ранних сроках беременности, особенно в период органогенеза.
Источник:
Elizabeth C. Ailes, April D. Summers, Emmy L. Tran, et al.
MMWR Morbidity and Mortality Weekly Report. 2018;67:18-22.
internist.ru
ТЯЖЕЛЫЙ ПОБОЧНЫЙ ЭФФЕКТ ДЕНОСУМАБА: ВРАЧИ ПРОТИВ ФАРМАЦЕВТИЧЕСКОЙ КОМПАНИИ
Швейцарские врачи бьют тревогу — популярный препарат против остеопороза деносумаб (Пролиа) после прекращения терапии может вызывать парадоксальный эффект отмены.
Сомнительная безопасность терапии
С 2015 года клиницисты стали подозревать, что деносумаб ассоциирован с появлением боли в костях и повышением риска спонтанных переломов позвонков после завершения курса терапии.
После отмены препарата у пациентов отмечается повышение маркеров костного ремоделирования и быстрое снижение минеральной плотности костной ткани.
Результаты исследований показывают, что после окончания терапии деносумабом костная резорбция быстро восстанавливается и ее активность в течение 12 месяцев после последней инъекции выше в 2 раза, чем до терапии.
Швейцарский фармакологический надзор – Swissmedic – продолжает расследование по безопасности терапии. Это уже нашло отражение: в 2017 году изменена инструкция к препарату Пролиа.
Комментарии разных сторон
Компания Amgen, производитель деносумаба, отказывается давать комментарии журналистам. Представители компании заявляют, что повышение риска переломов по окончанию терапии является для них большим сюрпризом. Как такое возможно, говорят они, ведь препарат призван бороться с остеопорозом, имеет большое преимущество перед другими лекарствами, так как вводится всего 1 раз в 6 месяцев.
А вот мнение специалистов в области клинической фармакологии прямо противоположно. Так, профессор Университетской Клиники в Лозанне Thierry BUCLIN отмечает, что это напоминает скандальную историю с противодиабетическим препаратом Медиатор, который использовался у пациентов с сахарным диабетом в качестве средства, уменьшающего массу тела. Препарат на протяжении длительного периода применялся во Франции, хотя на фоне терапии были неоднократно зафиксированы признаки вальвулопатии. После длительной тяжбы выяснилось, что основной компонент Медиатора относится к группе амфетаминов, которые являются сильнейшими психостимуляторами, что и объяснило большое число нежелательных реакций, в том числе и со стороны сердца.
Швейцарские врачи надеются, что в компании Amgen задумаются о безопасности терапии и проведут дополнительные исследования.
Источники:
Olivier Lamy, Elena Gonzalez-Rodriguez, Delphine Stoll, Bérengère Aubry-Rozier.
Rev Med Suisse 2017; volume 13. 863-866.
Juliette Galeazzi. L’ostéoporose au coeur d’un scandale découvert à Lausanne. www.rts.ch
internist
КАРФИЛЗОМИБ-АССОЦИИРОВАННЫЕ ПОБОЧНЫЕ ЭФФЕКТЫ, РЕЗУЛЬТАТЫ СИСТЕМАТИЧЕСКОГО ОБЗОРА И МЕТА-АНАЛИЗА
Карфилзомиб – препарат, используемый для лечения множественной миеломы, может вызывать жизнеугрожающие сердечно-сосудистые побочные эффекты.
Опубликован первый систематически обор и мета-анализ, выполненный для оценки сердечно-сосудистой безопасности препарата.
Методы
Исследователи провели анализ медицинских баз данных PubMed, EMBASE, Web of Science и clinicaltrials.gov с целью поиска клинических исследований 1-3 фазы.
Основной конечной точкой являлась частота сердечно-сосудистых событий (сердечная недостаточность, артериальная гипертензия, ишемия и аритмия). Ученые также оценивали степень тяжести нежелательных явлений.
Результаты
Исследователи отобрали 514 исследований, из которых 24 вошли в финальный анализ (2 594 пациента с множественной миеломой). Итоги анализа:
- Общее число нежелательных явлений составило 617 (18,1%), из которых 274 (8,2%) расценивались как побочные эффекты 3-й и более степени тяжести.
- Применение карфилзомиба в дозе 45 мг/м2 и выше было ассоциировано с более выраженной тяжестью побочных эффектов.
- Показано, что указание в анамнезе на предшествующую терапию и конкурентное применение препаратов для лечения множественной миеломы не были ассоциированы с частотой кардиоваскулярных нежелательных явлений.
- Анализ рандомизированных исследований показал, что относительный риск развития побочных эффектов любой степени тяжести составил 1,8, а риск нежелательных явлений 3 и большей степени – 2,2.
Заключение
Применение карфилзомиба ассоциировано с достоверным повышением частоты сердечно-сосудистых побочных эффектов, при этом риск носит дозозависимый эффект.
Источник:
Adam J. Waxman, Suparna Clasen, Wei-Ting Hwang, et al.
JAMA Oncol. Published online December 28, 2017.
Сочетание БАДов и лекарств может негативно отразиться на здоровье
Команда учёных из разных стран провела исследование, направленное на изучение сочетания травяных препаратов и обычных лекарств. В результате были описаны клинические случаи, когда пациенты в результате такого совмещения наносили вред здоровью.
Исследователи обнаружили множество описанных в медицине случаев, когда травяные препараты меняли эффект рецептурных лекарств: ослабляли, усиливали либо вызывали потенциально опасные побочные эффекты. Об этом сообщает the Guardian.
В число трав, которые могут оказать такое действие, вошли, в первую очередь, зверобой, женьшень и гинкго билоба.
К лекарствам, которые наиболее опасно сочетать с травяными средствами, специалисты отнесли антидепрессанты, препараты антиретровирусной терапии, статины, а также лекарства, применяемые при эпилепсии и сердечных заболеваниях, гормональные контрацептивы.
Руководитель исследования Чарльз АВОРТВЕ, учёный из Стелленбосского университета (Южная Африка), рассказал, что его команда выявила много клинических случаев, когда у пациентов наблюдались неблагоприятные реакции.
Так, описано несколько случаев по пациентам, имеющим проблемы с сердцем и принимающим варфарин или статины. У них были диагностированы осложнения, связанные с употреблением шалфея, льняного семени, зверобоя и зелёного чая.
«Если вы принимаете травяные средства, вы должны рассказать об этом своему врачу, – рекомендует Авортве. – Потенциальное взаимодействие и его последствия могут быть очень пагубными для здоровья пациента».
Ксения МАКСИМОВА
sibmeda.ru
ПРОИЗВОДИТЕЛЬ ПРИНЯЛ РЕШЕНИЕ ОТОЗВАТЬ ИЗ ОБРАЩЕНИЯ КОКЛЮШНО-ДИФТЕРИЙНО-СТОЛБНЯЧНУЮ ВАКЦИНУ (АКДС)

Производитель информирует об отзыве из обращения препарата «Вакцина коклюшно-дифтерийно-столбнячная адсорбированная (АКДС-вакцина), суспензия для внутримышечного введения, 0,5 мл/доза, ампулы (10), пачки картонные».
О принятом производителем решении сообщает Росздравнадзор. Речь идёт о серии препарата У53 производства ФГУП «НПО «Микроген» Минздрава России.
Причина таких мер – в несоответствии требованиям нормативной документации по показателю «Специфическая безопасность (коклюшного компонента)».
Росздравнадзор предписывает медицинским организациям предоставить в территориальные управления сведения, подтверждающие возврат указанной серии лекарственного препарата поставщикам либо производителю.
Ксения МАКСИМОВА
sibmeda.ru
ПРЕДУПРЕЖДЕНИЕ FDA О КОНТРАСТНОМ ВЕЩЕСТВЕ, СОДЕРЖАЩЕМ ГАДОЛИНИЙ
U.S. Food and Drug Administration (FDA) и Европейское Медицинское Агентство (ЕМА) ранее выпустили предупреждения относительно некоторых гадолиний-содержащих контрастных препаратов в связи с сообщениями об их накоплении в тканях головного мозга.
В декабре 2017 года FDA выпустило обновленные рекомендации, согласно которым все гадолиний-содержащие контрастные препараты, используемые во время МРТ для визуализации любых органов, могут накаливаться в головном мозге.
Такое скопление гадолиния может быть ассоциировано с прямым развитием побочных эффектов у пациентов, в том числе и с нормальной почечной функцией. По мнению экспертов, при этом польза от исследований продолжает превышать риски. Важно знать, что описаны случаи нефрогенного системного фиброза при задержке гадолиния у пациентов с предшествующей почечной недостаточностью.
В настоящем сообщении FDA отмечается необходимость проведения исследований на людях и животных с целью оценить безопасность данных продуктов.
Применение гадолиний-содержащих препаратов
Контрастные вещества, содержащие гадолиний, используется для визуализации опухолей, инфекционных очагов и поиска источника кровотечений. Гадолиний представляет собой тяжелый металл, большая часть которого выделяется с почками, однако следы вещества могут сохраняться в теле на протяжении длительного времени.
Важно выделить категории пациентов, у которых риск задержки гадолиния повышен. К ним можно отнести лиц, которым неоднократно выполняют исследования с гадолинием, беременных женщин, детей и пациентов с воспалением.
В настоящее время на рынке представлены 2 типа гадолиний-содержащих препаратов, линейные и макроциклические соединения. Необходимо отметить, что линейные вещества чаще и на более длительный срок задерживаются в организме.
До окончания анализа безопасности FDA рекомендует предупреждать всех пациентов о возможном риске гадолиний-содержащих контрастных веществ.
Источник:
FDA. Drug Safety and Availability. 19 December 2017
ГЕННАЯ ТЕРАПИЯ NOVARTIS ПОЛУЧИЛА УСКОРЕННОЕ РАССМОТРЕНИЕ В США И ЕС
Метод генной терапии Kymria, разработанный компанией Novartis, будет рассмотрен в ускоренном порядке регуляторами Европы и США в качестве метода лечения диффузной В-крупноклеточной лимфомы у взрослых.
Как сообщает Reuters, об этом производитель официально объявил 17 января текущего года. Ранее Kymria была одобрена в качестве метода лечения острого лимфобластного лейкоза у детей и подростков.
Благодаря ускоренной процедуре рассмотрения решение будет вынесено в США в течение 6 месяцев вместо 10, а в Европе – в течение 150 дней, тогда как стандартная процедура занимает 210.
В основе Kymria – использование собственных Т-клеток пациента, которые модифицируют, добавляя к ним химерный антигенный рецептор (Chimeric antigen receptor, CAR).
Этот CAR-рецептор направляет Т-клетки на борьбу с раковыми клетками, у которых есть B-лимфоцитарный антиген CD19 на поверхности. Таким образом, организм пациента становится способен сам бороться с лейкемией.
По предварительным прогнозам, при получении дополнительного одобрения продажи Kymria принесут компании более миллиарда долларов.
riaami.ru
КАЛЬЦИЕВЫЕ ДОБАВКИ: ВЛИЯНИЕ НА РИСК ПЕРЕЛОМОВ У ПОЖИЛЫХ ПАЦИЕНТОВ
Увеличение частоты переломов, ассоциированных с остеопорозом, является важной социальной и экономической проблемой. Результаты исследований, оценивающих эффект кальциевых добавок на риск переломов у пожилых пациентов, носят противоречивый характер, несмотря на это, многие врачи продолжают рекомендовать их прием.
Так есть ли польза от кальциевых добавок или они так же бесполезны как мультивитаминные комплексы? Ключом к ответу на данный вопрос служит мета-анализ.
Методы
Ученые использовали базы данных PubMed, Cochrane library и EMBASE для поиска рандомизированных клинических исследований (РКИ), в которых оценивали влияние добавок кальция на частоту переломов у лиц старше 50 лет. Это были исследования, опубликованные с июля 2012 года по июль 2017 года.
В качестве первичной конечной точки рассматривалась частота переломов бедра, вторичными конечными точками являлись общая частота переломов, частота вертебральных и периферических переломов.
Результаты
В мета-анализ вошли 14 РКИ, в которых сравнивали кальциевые добавки с плацебо или отсутствием терапии.
Показано, что применение кальциевых добавок не приводило к достоверному снижению риска переломов бедра (относительный риск, 1,53 [95% CI, 0,97-2,42], ARD, 0.01 [95% CI, 0,00-0.01]). При этом доза кальция (≥1 г или <1 г) не оказывала влияния на результат.
Не было выявлено статистически значимой ассоциации между приемом кальциевых добавок и периферическими переломами (относительный риск, 0,95 [95% CI, 0,82-1,11], ARD, –0,01 [95% CI, –0,02-0,01]), переломами позвоночника (относительный риск, 0,83 [95% CI, 0,66-1,05], ARD, –0,01 [95% CI, −0,03-0,01]) и общим риском переломов (относительный риск, 0,88 [95% CI, 0,75-1,03]; ARD, −0,02 [95% CI, −0,03- −0,01]) по сравнению с плацебо или отсутствием терапии как таковой.
Исследователи не выявили достоверных различий в подгруппах пациентов при учете дозы кальция, пола, предшествующих переломов, ежедневного потребления кальция с пищей и базового уровня 25-ОН-D.
Источник:
Jia-Guo Zhao, Xian-Tie Zeng, Jia Wang, et al. JAMA. 2017;318(24):2466-2482.
ПОЛУЧЕНЫ НОВЫЕ ДАННЫЕ ПО БЕЗОПАСНОСТИ ПРИМЕНЕНИЯ ПРЕПАРАТА «НАЗОЛ® БЭБИ»
Росздравнадзор сообщил о решении компании АО «Байер» внести изменения в данные по безопасности препарата «Назол® Бэби» относительно рекомендованного возраста его применения.
В письме, адресованном специалистам здравоохранения, компания АО «Байер» сообщает о получении данных по безопасности лекарственного препарата «Назол® Бэби» (фенилэфрин), капли назальные, 0,125% (NoPY: П N016016/01).
В связи с этим вносятся изменения в данные о рекомендованном возрасте применения этого лекарственного препарата.
Теперь возрастной порог применения лекарственного препарата устанавливается на уровне 2 месяцев вместо 0 месяцев в утверждённой на данный момент версии инструкции по медицинскому применению от 18.05.2016 г. В настоящее время компания проводит работу по включению изменений в инструкцию.
Ксения МАКСИМОВА
sibmeda.r
ПОПУЛЯРНЫЙ АНТИБИОТИК ВНОВЬ ИЗЫМАЮТ ИЗ-ЗА РАЗВИТИЯ ПОБОЧНОЙ РЕАКЦИИ
 Федеральная служба по надзору в сфере здравоохранения вновь сообщает об изъятии популярного в российских больницах антибиотика «Цефтриаксон». На этот раз изымается препарат производства ОАО «Биохимик».
Федеральная служба по надзору в сфере здравоохранения вновь сообщает об изъятии популярного в российских больницах антибиотика «Цефтриаксон». На этот раз изымается препарат производства ОАО «Биохимик».
Российская фармкомпания «Биохимик» решила приостановить реализацию препарата «Цефтриаксон, порошок для приготовления раствора для внутривенного и внутримышечного введения 1,0 г, флаконы (50), коробки картонные (для стационара)» серии 241015. Такое решение принято в связи с развитием нежелательной реакции.
Росздравнадзор просит медицинские организации проверить наличие указанной серии препарата и прекратить ее использование. Антибиотик будет изъят из обращения.
За последние два месяца текущего года это уже третье изъятие «Цефтриаксона» из-за развития нежелательной реакции. В первые два раза изымался антибиотик другого производителя – ООО «КОМПАНИЯ «ДЕКО».
«Цефтриаксон» – самый популярный антибиотик в российских больницах, он входит в ТОП-10 по объемам закупок лекарственных средств лечебными учреждениям.
clinical-pharmacy.ru
FDA не одобрило препарат Олюмиант (барицитиниб) для лечения ревматоидного артрита
 FDA не утвердило препарат Олюмиант/Olumiant (барицитиниб / baricitinib) компаний Eli Lilly и Incyte, предназначенный для лечения умеренной и тяжелой формы ревматоидного артрита. Регулятор потребовал предоставить дополнительные данные касательно безопасности лекарственного средства и наиболее приемлемых дозировках.
FDA не утвердило препарат Олюмиант/Olumiant (барицитиниб / baricitinib) компаний Eli Lilly и Incyte, предназначенный для лечения умеренной и тяжелой формы ревматоидного артрита. Регулятор потребовал предоставить дополнительные данные касательно безопасности лекарственного средства и наиболее приемлемых дозировках.
Как отметили в Eli Lilly, компания уверена в безопасности барицитиниба и не согласна с решением FDA. Разработчики препарата планируют продолжить сотрудничество с регуляторным органом и будут подавать повторную заявку на его регистрацию.
Препарат Олюмиант (барицитиниб) уже одобрен в Европе для лечения умеренной и тяжелой формы ревматоидного артрита.
clinical-pharmacy.ru
АНТИБИОТИКИ СВЯЗАЛИ С ВЫСОКИМ РИСКОМ ВЫКИДЫША
Макролиды, хинолоны, тетрациклины, сульфонамиды и метронидазол связали с повышенным риском самопроизвольного аборта, то есть потери беременности до двадцатой недели, говорится в исследовании, опубликованном в журнале Канадской медицинской ассоциации.
Исследование было проведено в Квебеке. Взяли 8 700 случаев самопроизвольного аборта и 87 000 успешных беременностей в качестве контрольной группы, и изучали медицинские назначения, сделанные матерям в возрасте от 15 до 45 лет. Работа с этой статистикой была возможна потому, что все пациенты были застрахованы в местной системе обязательного медицинского страхования, покрывающей и расходы на лекарство (следовательно, вся отчетность тоже хранится годами).
Выяснилось, что примерно 16% матерей, женщин, пострадавших от выкидыша, принимали антибиотики в первой половине беременности, и только 13% матерей, чья беременность завершилась родами. Антибиотик азитромицин увеличивал шанс выкидыша на 65%, кларитромицин в два раза. Норфлаксацин увеличивал риск в 4 раза.
Как и многие другие подобные исследования, которые ищут корреляции между разными явлениями и процессами, само по себе оно ничего не доказывает. Большая часть выкидышей происходит по генетическим причинам. Тем не менее, масштаб исследования и серьезность корреляций по крайне мере требует новых исследований и определенной настороженности в назначении антибиотиков. Авторы исследования говорят, что удивились, узнав, какому большому количеству беременных женщин назначают такие препараты.
Тем не менее, в ряде случаев этого избежать нельзя, и при бактериальной инфекции беременным все равно нужно назначать антибиотики, правда, в минимально возможных эффективных дозах.
medportal.ru
В ЕВРОПЕ РЕКОМЕНДОВАНО К ОДОБРЕНИЮ 11 ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ
Комитет по лекарственным средствам для медицинского применения (Committee for Medicinal Products for Human Use – CHMP) Европейского агентства по лекарственным средствам (European Medicines Agency – EMA рекомендовал к одобрению 11 препаратов, в том числе 4 орфанных и 3 биосимиляра.
Эксперты СНМР рекомендовали зарегистрировать 2 орфанных препарата для применения у детей. Это Спинраза/Spinraza (нусинерсен/nusinersen) для терапии спинальной мышечной атрофии и Бринеура/Brineura (церлипоназа альфа/cerliponase alfa) для лечения нейронального цероид-липофусциноза типа 2.
Также был рекомендован к одобрению препарат Беспонса/Besponsa (инотузумаб озогамицин/inotuzumab ozogamicin) для лечения острого лимфобластного лейкоза, который также имеет статус орфанного препарата.
Положительную оценку эксперты дали лекарственному средству Куприор/Cuprior (триантин тетрагидрохлорид/trientine tetrahydrochloride), предназначенному для лечения болезни Вильсона, редкого аутосомно-рецессивного наследственного заболевания. Препарат имеет также статус орфанного.
СНМР рекомендовал к одобрению препарат Кевзара/Kevzara (сарилумаб/sarilumab), предназначенный для лечения ревматоидного артрита, и препарат Скиларенс/Skilarence (диметилфумарат/dimethyl fumarate) для лечения псориаза.
Эксперты рекомендовали расширить показания к применению трех лекарственных средств: Авастин (бевацизумаб), Целсентри (маравирок) и Опдиво (ниволумаб).
СНМР одобрил три биосимиляра: Эрелзи (этанерцепт), Риксатон и Риксимио (ритуксимаб).
clinical-pharmacy.ru
РИСК НЕЙРОДЕГЕНЕРАТИВНЫХ ЗАБОЛЕВАНИЙ У ПАЦИЕНТОВ, ПОЛУЧАЮЩИХ МЕТФОРМИН
В Женеве, на прошедшей в апреле 2017 года международной конференции по болезни Альцгеймера и болезни Паркинсона были представлены результаты исследования, показавшего, что у пациентов с сахарным диабетом, получающих метформин, повышен риск развития нейродегенеративных заболеваний.
Методы. В когортное исследование были включены 9 302 пациента с СД 2 типом из Тайваня. 4 651 пациент получал метформин и 4 651 пациент не получал данный препарат (группа контроля). У пациентов оценивали заболеваемость болезнью Альцгеймера и Паркинсона на протяжении 12 лет с поправкой на возраст, пол, тяжесть сахарного диабета.
Результаты. Анализ продемонстрировал, что частота развития болезни Паркинсона составила 6,85% в группе метформина и 2.78% в группе контроля (скорректированный коэффициент рисков, 2.27 (95% CI 1.66-3.07), частота деменции любой этиологии была равна 11,5% и 6.7% соответственно (коэффициент рисков, 1.66 (95% CI 1.35-2.04).
Болезнь Альцгеймера была диагностирована у 1,64% в группе метформина и у 0,83% больных из контрольной группы (коэффициент рисков, 2.13 (95% CI 1.20-3.79). Сосудистая деменция была выявлена у 1,64% и 0,69% соответственно (коэффициент рисков, 2.30 (95% CI 1.25-4.22). Обращало на себя внимание повышение частоты заболеваний при продолжительной терапии метформином более 300 дней и в дозе более 240 г.
Риск болезни Паркинсона достоверно повышался, начиная с продолжительности приема метформина на протяжении более 300 дней: 300-400 дней – 2.20 (1.47-3.28), ≥400 дней – коэффициент рисков составил 4.49 (3.06-6.58). Для деменции любой этиологии риск достоверно увеличивался при периоде лечения 300-400 дней (1.61; 1.21-2.16) и длительности более 400 дней (2.84; 2.12-3.82). Выявлено дозозависимое повышение риска болезни Паркинсона. У пациентов, получавших 240-385 г препарата, коэффициент рисков составил 2.14 (1.41-3.25), у лиц, получавших >385 г – 3.54 (2.41-5.20). Такой же дозозависимый эффект наблюдался в отношении повышения риска деменции. Коэффициент рисков для 240-383 г составил 2.06 (1.54-2.75), для дозы более 385 г – 1.97 (1.45-2.68).
Обсуждение. По мнению некоторых экспертов, результаты одновременно удивительны и вызывают много вопросов. К примеру, не было ли различия уровня гликированного гемоглобина между группами или значительного различия между дополнительными противодиабетическими препаратами, что может повлиять на результаты исследования.
Источник:
AD/PD 2017:
International Conference
on Alzheimer’s and Parkinson’s Diseases.
Abstract 312
ПРОТИВОЭПИЛЕПТИЧЕСКИЕ ПРЕПАРАТЫ ПРИВЕЛИ К ВРОЖДЕННЫМ ПОРОКАМ 4 ТЫСЯЧ ДЕТЕЙ ВО ФРАНЦИИ
Во Франции от 2,15 до 4,1 тысячи детей с 1976 по 2016 годы родились с серьезным врожденным пороком из-за того, что при беременности их матери принимали лекарственные препараты от эпилепсии с вальпроевой кислотой, следует из исследования Национального агентства по безопасности лекарственных средств и товаров для здоровья Франции (ANSM).
Препараты продавались на нескольких мировых рынках, включая США, Китай, Францию и Великобританию. Некоторые из этих препаратов (например, Depakine) выпускала французская компания Sanofi, отмечает газета Financial Times.
Тератогенные эффекты (вызывающие нарушения в эмбриональном развитии) вальпроевой кислоты были известны с начала 1980-х годов. Совсем недавно, в 2000-е годы, повышенный риск задержки развития и расстройств аутистического спектра был выявлен у детей, подвергшихся воздействию вальпроевой кислоты в утробе матери», – говорится в докладе ANSM.
Также отмечается, что риск возникновения серьезных врожденных пороков возрастает в четыре раза, если мать принимает вальпроевую кислоту при лечении эпилепсии, и в два раза, если мать принимает препарат для лечения биполярного расстройства.
Компания Sanofi с 2011 года предупреждала, что препарат не следует использовать во время беременности. «Мы знаем о ситуациях в семьях, где у детей есть проблемы, которые могут быть связаны с использованием противоэпилептических препаратов их матерями при беременности», – приводит издание заявление компании.
1prime.ru
В ЕВРОПЕ УТВЕРЖДЕН НОВЫЙ АЛГОРИТМ РЕАГИРОВАНИЯ НА НАРУШЕНИЕ НАДЛЕЖАЩИХ ПРАКТИК
Европейское агентство по лекарственным средствам (EMA) утвердило новый алгоритм реагирования на заявления о нарушениях надлежащих фармацевтических практик, присылаемые сторонними наблюдателями.
Теперь сообщения любого работника предприятия о несоблюдении стандартов GMP при выпуске лекарств смогут повлечь за собой проверку завода-производителя и принятие дополнительных контрольных мер по отношению к лекарственному препарату.
Целью нововведений было создание доверительной атмосферы, благодаря которой любой мог бы изложить свои наблюдения о нарушениях. Новая политика позволит EMA оценивать поступающие сообщения и координировать дальнейшие расследования инцидентов, сохраняя при этом конфиденциальность источника.
С 2013 года в EMA поступило 43 сообщения о нарушениях на производстве или при проведении клинических исследований, хотя до настоящего времени не было специального алгоритма реагирования на подобные сообщения.
Для индивидуальных обращений в EMA по поводу выявленных нарушений была создана электронная почта: reporting@ema.europa.eu. В агентстве подчеркивают, что по заявлениям будет проведена соответствующая проверка с принятием необходимых мер по устранению нарушений.
pharm.reviews
МИНЗДРАВ США РАЗРЕШИЛ ПРИМЕНЯТЬ НОВЕЙШИЕ ПРОТИВОВИРУСНЫЕ ПРЕПАРАТЫ ДЛЯ ТЕРАПИИ ГЕПАТИТА С И ПРИ ЛЕЧЕНИИ ДЕТЕЙ
Решением надзорного ведомства при Минздраве США изменены возрастные ограничения для применения противогепатитных препаратов софосбувир и комбинации софосбувир+ледипасвиром. Теперь эти лекарственные средства можно применять для лечения HCV-инфекции и у пациентов в возрасте от 12 до 17 лет.
Управление по контролю качества пищевых продуктов и лекарственных препаратов США (FDA) расширило область применения препаратов Совальди (софосбувир) и Хармони (софосбувир+ледипасвир).
Теперь эти лекарственные средства, созданные учеными из фармацевтической компании Gilead Sciences, Inc., можно применять для лечения вирусного гепатита С 6 главных генотипов у пациентов с 12 лет. Ранее оба лекарственных средства были зарегистрированы в США для лечения этой формы гепатита у взрослых.
Совальди (софосбувир) теперь можно назначать пациентам детского возраста, начиная с 12 лет, для лечения HCV-инфекции и в комбинации с рибавирином.
Условием для назначения новых препаратов является масса тела пациента не менее 35 кг и отсутствие тяжелого цирроза печени, допускается применение этих лекарственных средств при лечении вирусного гепатита С у пациентов с циррозом умеренной степени тяжести.
По оценке специалистов Центров по контролю заболеваний и их профилактике (Centers for Disease Control and Prevention) в настоящее время в США число детей в возрасте до 18 лет, страдающих HCV-инфекцией, оценивается в 23 000-46 000 человек.
fda.gov
NICE НЕ РЕКОМЕНДОВАЛ ПРЕПАРАТ ДАРЗАЛЕКС (ДАРАТУМУМАБ) ДЛЯ ЛЕЧЕНИЯ МНОЖЕСТВЕННОЙ МИЕЛОМЫ
Национальный институт здоровья Великобритании (NICE)не одобрил применение препарата дарзалекс/darzalex (даратумумаб /daratumumab) американской компании Johnson & Johnson, предназначенного для лечения множественной миеломы.
Эксперты, рассмотрев предоставленные компанией данные по эффективности применения лекарственного средства у пациентов, которым не подошли другие лекарственные средства, не признали Дарзалекс соответствующим стандартам соотношения стоимости и эффективности. Таким образом, институт NICE не рекомендовал использование препарата в качестве монотерапии у пациентов с множественной миеломой.
В компании Johnson & Johnson обеспокоены решением NICE. Если оно окажется окончательным, то пациенты с множественной миеломой, которым не подошли одобренные методы терапии, не смогут получить доступ к инновационному препарату.
Весной прошлого года Еврокомиссия зарегистрировала лекарственный препарат «Дарзалекс» (даратумумаб) для применения у пациентов с рецидивирующей или рефрактерной множественной миеломой, у которых заболевание прогрессировало после терапии на основе ингибиторов протеасом и иммуномодуляторов.
Основой для одобрения лекарственного средства стали результаты клинических исследований, согласно которым медиана общей выживаемости составила 20 месяцев у пациентов, получавших лекарственный препарат «Даратумумаб» в качестве средства монотерапии. Общая частота ответов составила 31%, примерно у 83% пациентов было зафиксировано улучшение состояния.
healtheconomics.ru
FDA НЕ ОДОБРИЛО ПРЕПАРАТ ОЛЮМИАНТ (БАРИЦИТИНИБ) ДЛЯ ЛЕЧЕНИЯ РЕВМАТОИДНОГО АРТРИТА
FDA не утвердило препарат Олюмиант/Olumiant (барицитиниб / baricitinib) компаний Eli Lilly и Incyte, предназначенный для лечения умеренной и тяжелой формы ревматоидного артрита.
Регулятор потребовал предоставить дополнительные данные касательно безопасности лекарственного средства и наиболее приемлемых дозировках.
Как отметили в Eli Lilly, компания уверена в безопасности барицитиниба и не согласна с решением FDA. Разработчики препарата планируют продолжить сотрудничество с регуляторным органом и будут подавать повторную заявку на его регистрацию.
Олюмиант (барицитиниб) уже одобрен в Европе для лечения умеренной и тяжелой формы ревматоидного артрита.
clinical-pharmacy.ru